![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
6.11 Computers in Nucleic Acid Sequencing
The spectacular strides in nucleic acid sequencing have, in recent years, led to an exponential growth of knowledge about the primary structure of various nucleic acids. If sequences containing a total of at least a million nucleotides had been established by 1983, by the year 1985 this number had increased to four million, by September of 1986 - to nine million, and by the end of 1987 - to about ten million. As has already been mentioned, determination of the primary structure of the human genome (3 billion nucleotide pairs) is already being discussed today. In addition to automatic DNA sequencers, accomplishment of this task calls for powerful computers and software for data acquisition, storage and analysis. The extraordinary progress achieved over the past few years in microelectronics has led to the advent of mini- and microcomputers of the IBM PC type which, by virtue of their broad potential (memory size, speed, low cost and, as a result, ready availability to laboratories and even individual workers), have made computer analysis of the genome structure and functions routine for thousands of molecular biologists without any programming and computer background but capable of tackling any problem related to the functioning of biomolecules with the aid of computers. There is every reason to consider the late eighties as the dawn of the computer age in molecular biology. The possibility of producing "nucleotide texts" which encode genetic information provides new tools for its interpretation. What this means is new approaches that can be instrumental in finding not only loci of structural genes in any genetic information (since the protein code is known) but also regulation sites (promoters, operators, enhancers, splicing sites, etc.). All this is done using the information theory and statistical methods as well as such research tools as computers, nucleotide sequence data bases, and dedicated software packages.
Let us now return to the project aimed at establishing the primary structure of the human genome in its entirety. Remarkably, gene sequences account for only five to ten per cent of the genome. Knowledge of the primary structure of the entire genome (plus finding out what is encoded in the remaining sequences) opens up new possibilities in elucidating the genetic contribution to all aspects of normal and abnormal functioning of the living organism. Comparison of the findings with primary genome structures of other organisms is of greatest interest from the standpoint of evolution of the encoded genes and macromolecules. This may also be helpful in learning the language whereby the human gene expression is controlled. Since computers can store any amount of data on the primary genome structure and easily access the necessary nucleotide sequences, one can rapidly accomplish such tasks as determination of the primary structure of the protein encoded in the gene, comparison of the primary structures of genes (and, consequently, proteins), and identification of translation frames.
The most important application of computers has to do with the prediction of three-dimensional structures of ribonucleic acids and proteins. For instance, this approach enables one to predict three-dimensional structures of DNA introns acting as enzymes. It is widely used in gene engineering for proper selection of amino acid replacements.
The need to solve all these problems has led to the development of special centers for acquiring data on the primary nucleic acid structure as well as commercial dedicated software packages for complete analysis of such data.
Ale early stages of DNA and RNA sequencing were already marked by setting up of international systems for data storage. The most famous data banks of nucleotide sequences are located in the United States (Gene Bank established in 1982 on the basis of the Los Alamos data base) and in Europe (EBML bank created in Heidelberg in 1980). In the Soviet Union, the AllUnion Bank of Nucleotide Sequences has been established under the auspices of the USSR Academy of Sciences. These and similar organizations specialize in collection of the published nucleotide sequences and releasing of the available information. The activities of such centers also include the following: data reduction and creation of directories; entering, editing and storage of primary structure data; and systematization of sequences, based on different parameters. The created data retrieval systems ensure rapid access to any sequence or accomplishment of any of the above tasks. The information is made available to users either on magnetic carriers or in the form of printouts or catalogues. The astounding rate of acquisition of DNA and RNA sequences is already challenging designers to find ways of speeding up their transmission to data banks and compressing the data for storage without adversely affecting their accessibility. Revolutionary changes are to be expected in the nearest future not only in experimental sequencing methods but also in the hardware, primarily development of new types of computers and their operating systems, wide-spread use of optical storage devices (CDROMS), and elaboration of electronic methods for data acquisition and distribution.


Fig. 6-37. Form sent to the Gene Bank (USA).
Proposals are being made to send all newly decoded sequences directly to data banks without their publication in journals (to avoid errors arising from reprinting). It has been recommended to transmit data by telephone links. New forms of presenting nucleotide sequences have been developed, which lend themselves to automatic reading by computers; that is, not in the four-letter form but in binary code, in a graphic form (as four vectors differently oriented in two or three dimensions), and even as audio signals (of four different tones). At present, more or less universal rules govern the format in which sequences must be sent to data banks. Figure 6-37 illustrates a typical form filled out prior to sending to the Gene Bank (USA). Symbols and the corresponding codes have been developed for designating nucleotides, including minor ones. In addition to data acquisition and storage, computers perform another major function which is analysis of nucleic acid sequences. Today, a large number of software products (usually in the form of packages) are available for this purpose. They free research workers of many tedious routine operations, such as counting mono-, di- and trinucleotides, translation of a nucleotide sequence into that of amino acids, search for particular sites, comparison of sequences, and so on. Already in the early eighties, special issues of "Nuclear Acids Research" described software for storage and analysis of data on the primary structure of nucleic acids. All currently available software packages are tailored to particular computers, (users buy programs depending on the computer model at their disposal), and differ both in design and in the range of functions they can perform. Some of them include data from the Gene Bank or EBML. For example, four packages are commercially available at present for IBM PC or compatibles: DNASIS, DNASTAR, IBI, and MICROGENIE. The most popular software package in the Soviet Union was SEQBUS developed for PC "Iskra 226" at the Institute of Molecular Biology, USSR Academy of Sciences.
All programs can be conventionally divided into two categories: general purpose and special or dedicated. The former are intended for the most common operations of data acquisition and analysis and perform the following functions: entering and editing of new sequences; direct reading of autoradiograms and gels with the aid of scanning devices; location of restriction endonuclease recognition sites and presentation of data in a convenient (tabular or graphic) form; location of sites with components of rotational and mirror symmetry (palindromes); translation of a nucleotide sequence into a protein one in all three reading frames; comparison of two sequences by the homology dot matrix method; comparison of a new sequence with all those in the Gene Bank; location of sites rich in particular nucleotides; calculation of a hypothetical DNA melting point; automatic assembly of sequenced fragments into a single structure (DNA molecule); translation of a protein sequence into a nucleotide one with due account for irregular usage of synonymous codons; determination of the molecular weight of nucleic acids and proteins; prediction of the secondary protein structure; calculation of the free energy of hairpin formation; and many others.
Dedicated programs are developed to solve special, often more complicated problems and are of interest to a narrower group of specialists. They can perform e.g. a calculation of the length of DNA fragments on the basis of their electrophoretic mobility in gels; selection of hybridization probes; prediction of the secondary RNA structure; location of nucleotides in a gene, which can be altered (without changing the amino acid sequence) in order to introduce a restrictase recognition site; location of sites with a potentially possible Z-form of DNA structure; identification of functionally significant sites in an unknown newly decoded structure, based on a previous consensus (as a result of analysis of known structures performing the same function); location of protein-encoding sites; and so on. Figure 6-38 shows, by way of example, the menu of the MICROGENIE package to illustrate the general or special functions that can be chosen by the researcher when handling nucleotide sequences. The structure and functions of some programs will be covered at greater length in what follows.
The general-purpose program COMMON allows the user to enter a sequence into the computer and verify the entered data in an interactive

Fig. 6-38. Menu of MICROGENIE software package.
mode. Other possibilities offered by the program include deletions and inserts as well as elimination of sequences to simulate hybrid and mutant DNA molecules. Sequences are usually entered manually using a keyboard, but in recent years instruments have been developed for automatic scanning of autoradiograms and sequencing gels, obtained with the use of fluorescent labels, and direct data transmission to the computer with subsequent analysis of the sequence using special software. The book "Sequencing Analysis of Nucleotide Acids and Proteins", published recently by IRL Press (Oxford), contains detailed descriptions of both scanning devices and software for reading autoradiograms. Programs have been written for determination of the primary structures of high-molecular weight DNAs based on the fragment sequencing data obtained during non-specific cleavage (e.g., by sonication). Whatever relationship exists between any two DNA fragments becomes known from coincidence of the nucleotide sequences in their structures, the coinciding sequences serving as overlapping sites, and the two fragments can be reunited in a more extended structure. The process of fragment selection and joining continues as long as it takes to determine the entire primary structure in the DNA under analysis. One such program is CONTIG (with a modification for IBM PC), created at F. Sanger's laboratory (Cambridge, England). Here are the basic operations that can be executed using CONTIG:
(1) Storage of sequences for each fragment
(2) Selection of adjacent fragments to assemble sequences
(3) Comparison of data obtained from reading of new autoradiograms with already established sequences
(4) Joining of two fragments with the aid of a third one which represents the site where the first two overlap
(5) Search for DNA sites complementary to the already established ones, which serves as a check ensuring that the full DNA structure has been assembled correctly.
After the above-described reconstruction of the DNA a check is carried out using the data on restriction sites in the starting DNA, obtained while preparing a restriction map. Coincidence of the restriction site location data (this conclusion is drawn from prediction of the DNA fragmentation picture, based on the established primary structure of the DNA) indicates that the primary structure of the DNA under investigation has been determined correctly. Testimony to the enormous potential of computers in solving such problems is provided by the fact, for example, that data from 12 gels, each containing 200 to 300 nucleotides, can be assembled automatically into a continuous sequence within just a few seconds.
Almost every software package includes a program for identifying sites recognized by restriction endonucleases. Each package usually has a full list of the known restriction endonucleases (at present more than 450), and some packages also include a special sublist of only commercially available enzymes (about 100). One of such programs, SITOR, allows the user to construct a restriction map and print out a table of fragments of a DNA restricted at a predetermined site. The table contains the length and molecular weight of each fragment. The corresponding autoradiogram can be synthesized and presented graphically. Figure 6-39 illustrates the search for one of the restriction sites with the aid of the SITOR program.
Data banks provide the foundation on which all research with nucleotide sequences, aimed at determining the biological importance of individual DNA fragments, is based. Such research comprises two phases. The first phase involves grouping and subsequent comparative analysis (using appropriate criteria) of sequences performing the same biological function.
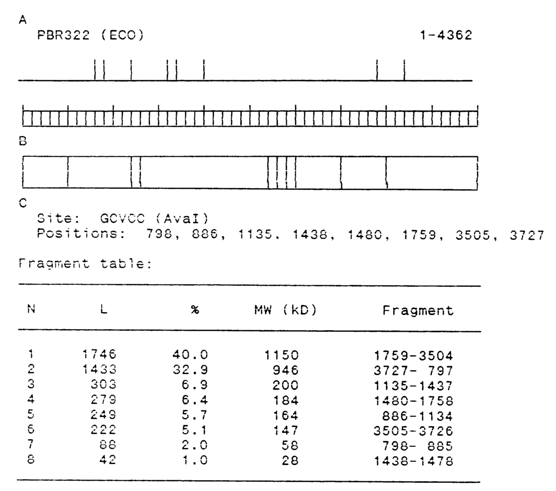
Fig. 6-39. Search for pentanucleotide GG(A/T)CC recognized by restrictase AvaI in plasmid pBR322. A - restriction map; B - autoradiogram; C - fragment table.
More specifically, it includes identification of "consensuses" (generalized statistically average structures) at regulatory sites of DNA (promoters, operators, enhancers, splicing sites, and ribosome recognition sites) or determination of the frequency of occurrence of synonymous codons in genes encoding different groups of proteins. The second, more interesting phase includes attempts to identify, within an unknown (newly decoded) structure, sites performing a particular function; for instance, search, based on homology with previously established consensuses, for gene-controlling sequences or potential protein-coding sites. Predictions of such DNA properties can be verified experimentally. The result may be a catalogue of properties of investigated DNAs.
The following programs may be mentioned as examples of software intended to determine the properties of particular sequences. One of the computer programs NAQ, for instance, has made it possible to establish the frequency of occurrence of codons in nucleotide sequences from yeast mitochondria, which encode proteins and do not do so, respectively. The resulting table for using codons later enabled investigators to identify polypeptide-encoding areas in unknown portions of the mitochondrial genome. Similar problems can be solved by means of the program PEPTIDOR. It permits the user to recognize opening reading frames on both DNA strands or, in other words, DNA portions encoding potential proteins from the arrangement of chain initiation and termination codons; to print out the amino acid sequence in the gene; to calculate the codon frequency; and to graphically represent the position of initiation and termination codons as well as the corresponding polypeptides. Figure 6-40 shows the distribution of encoded peptides in plasmid pBR322, the upward streaks standing for initiation codons, the downward streaks standing for termination codons, and the horizontal line standing for open reading frame.
It should be pointed out that to solve a more complex problem, such as identification of real protein-encoding sites among the potential ones, requires more sophisticated programs belonging to the category of dedicated software. They should also permit the user to perform more extensive analysis including search, near the boundaries of open frames, for so-called sequence signals homologous to regulatory sites (promoters, terminators, ribosome binding sites) typical of a particular organism, as well as analysis of the frequency of using synonymous codons within open reading frames and comparison with the data for known genes of the same organism (it has already been firmly established that the use of synonymous codons by all organisms is irregular, and this property can be used for identifying the encoding region for the organism in question). In the case of eukaryotic genes, the situation is complicated further due to the presence of introns. Yet the right approaches are already being worked out for them as well. A case in point is the program "ANALYSEQ" developed at Sanger's laboratory (it can be run on any graphic terminal) has such capabilities as search for sites encoding real proteins in both prokaryotes and eukaryotes.

Fig. 6-40. Map of possible polypeptides encoded in plasmid pBR322
An original approach to identification of functionally active sites is built in the program SASIP. It analyzes the frequency of octanucleotides in the Gene Bank sequences and generates the corresponding tables. According to postulates of the information theory, the most rarely occurring oligomers may perform the most important function. Comparison of the tabulated data with structures of fragments with known properties allows extremely important biological conclusions to be drawn as regards the structure and function of the genome.
Among the most important problems, (from the standpoint of understanding the molecular mechanism of biopolymer functions), which can be solved only with the aid of computers is construction of secondary RNA structures.
The currently existing programs, however smart they may be, are far from being perfect. But computer industry since it was established has been making so incredible progress that software producing companies have to do their best to stay in flow, and more powerful and sophisticated software packages are about to be released in the near future.
The development of fast DNA sequencing techniques gives a major impetus to molecular biology; it allows one, among other things, to elucidate the molecular mechanisms of the vital activity of organisms and their evolution.
Unfortunately, the rate of decoding sequences of nucleic acids lags far behind the current demand in analysis of their various fragments; each year, 106 bases are received by data banks. This is too little if it is remembered that the human genome alone contains 3.109 base pairs. Ways to speed up the reading of nucleic acid sequences should be sought in automation of the entire process, which will also free thousands of skilled operators from tedious work. Automation will be justified if the speed of analysis increases substantially (by several orders of magnitude) and its cost goes way down.
A prerequisite for solving the sequencing automation problem is availability of methods based on the simple principle of statistical degradation or biosynthesis. If in combination with such methods chemists and engineers pool their efforts, the problem of DNA and RNA sequencing may be solved in such a manner that before this century is over scientists will raise the veil of secrecy hiding not only the human genome but also that of other organisms.