![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
4.6 Properties of Nucleotide Derivatives with Substituents in the Phosphate Group
Advances in methods for analysing and synthesizing nucleic acids as well as studies into the mechanism of action of nucleotide metabolism enzymes have in recent years brought the properties of the phosphate group in nucleotides substituted at the phosphorus atom to the fore. This is why we have decided to devote this section to the properties of some compounds belonging to this category.
4.6.1 Nucleoside Cyclic Phosphates
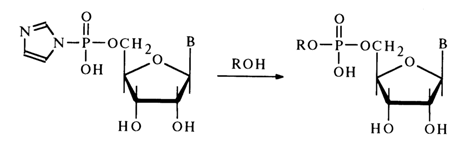
Nucleoside cyclic phosphates comprise an important class of nucleotide derivatives with respect to the phosphate group. They can be regarded as internal diesters of nucleosides. As has already been pointed out, five-membered ribonucleoside 2',3’-cyclic phosphates (23) are formed when RNA is dissociated under conditions of chemical or enzymatic hydrolysis and also as intermediate compounds during isomerization of ribonucleoside 2’- and 3’-phosphates in an acid medium. The methods for synthesizing these compounds were described in section 4.5.4.
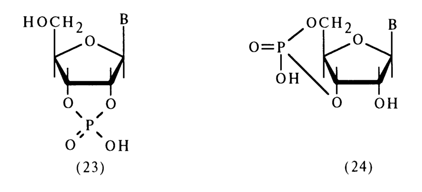
The cell has also been found to contain a six-membered cyclic phosphate, namely, adenosine 3',5'-cyclic phosphate [ belonging to compounds of type (24)]; it exists in a free state and play s a key role in the regulation of many biochemical processes.
Hydrolysis. Similarly to simple five-membered cyclic phosphates, ribonueleoside 2',3'-cyclic phosphates are highly labile compounds and can exist only at neutral pH values and low temperatures. They readily undergo hydrolysis in alkaline and acid media to yield nucleoside 2’- and 3’-phosphates in practically equal amounts:
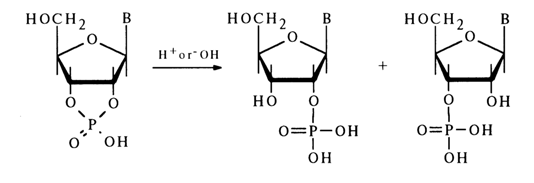
In an acid medium, hydrolysis of the P-O bond in nucleoside 2',3’-cyclic phosphates proceeds at a rate 106 to 107 times faster than in the case of simple dialkyl phosphates.
The reaction is reversible, although the rate of the backward process is much slower. A certain influence on the hydrolysis rate is exerted by the nature of the nucleotide's constituent base. Pyrimidine nucleoside 2',3’-cyclic phosphates are hydrolysed faster than the purine ones; the most labile compound is uridine 2',3’-cyclic phosphate. Alkaline hydrolysis of ribonucleoside 2',3’-cyclic phosphates is slower than acid hydrolysis, the reaction in this case being irreversible:
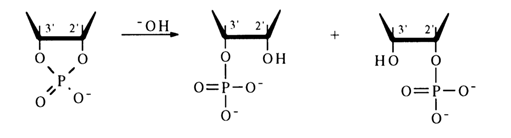
The greatest lability in an alkaline medium is displayed by uridine 2',3’-cyclic phosphate, and the greatest stability, by adenosine 2',3'-cyclic phosphate.
In weakly acidic and neutral media at 1000C, nucleoside 2',3’-cyclic phosphates are converted into the corresponding nucleosides. The reaction rate-determining step is hydrolysis of the phosphodiester bond, followed by rapid hydrolysis of the phosphomonoester one.
The ease of cleavage of the five-membered cycle in ribonucleoside 2',3’cyclic phosphates seems to be due (by analogy with simpler five-membered cyclic phosphates) to the ring being less strained in the transition state.
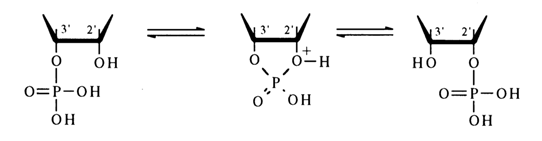
The formation of equal quantities of nucleoside 2’- and 3’-phosphates during acid as well as alkaline hydrolysis can be explained by the reaction passing with the same probability through two transition states differing in the nature of the hydroxyl which occupies an axial position (2' or 3’); in acid hydrolysis, any of the transition states readily undergoes pseudorotation.
Just as would be expected from comparison of the stabilities of five- and six-membered cyclic phosphates or simple glycols, ribonucleoside 3',5’- and deoxyribonucleoside 3',5'-cyclic phosphates are much less labile than the 2',3’isomers. As a rule, cleavage of the P-O bond during acid hydrolysis [path (a)] is accompanied by that of the glycosidic bond [path (b)]:
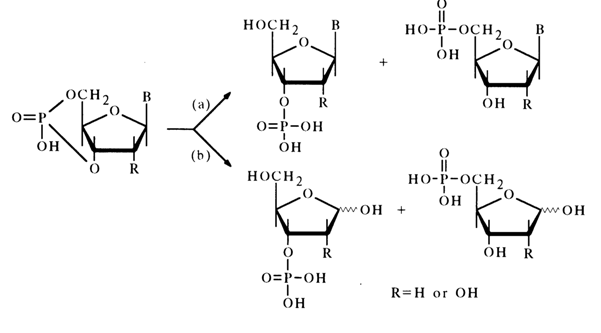
Hydrolysis along path (a) yields a mixture of nucleoside 3’- and 5’-phosphates (with the 3'-isomer being predominant), while hydrolysis along path (b) gives a mixture of the corresponding ribose or deoxyribose phosphates.
The main products of alkaline hydrolysis of nucleoside 3',5’-cyclic phosphates are nucleoside 3- and 5-phosphates forming at a 5:1 ratio [path (a)]. This process is particularly smooth in the presence of barium hydroxide, which seems to stem from the catalytic effect of barium ions capable of coordinating with the phosphate group.
Significantly, the rate of hydrolysis of the phosphodiester bonds in the sixmembered ring of nucleoside 3',5’-cyclic phosphate is still higher than in the case of ordinary six-membered cyclic phosphates, such as propanediol 1,3cyclic phosphate. This has to do with the peculiarities of the conformation of nucleoside 3',5’-cyclic phosphates in which the six-membered ring is trans-fused with the five-membered ribo- or deoxyribofuranose ring. X-ray structural analysis has shown that adenosine 3',5’-cyclic phosphate (25) has a structure with an unusual conformation of ribose in which C4 does not lie in the ring plane.
In the case of uridine 3',5-cyclic phosphate (26), a conformation of an envelope inwardly bent at C3, of ribose is assumed.
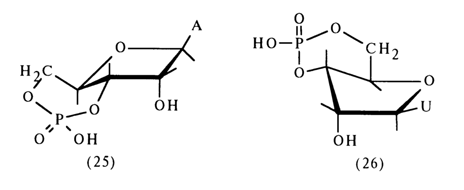
It is most likely that the faster cleavage of the P-O bond (at C5') in these compounds is due to the preferential formation (by virtue of the special conformational features of the nucleoside 3',5-cyclic phosphate structure) of such a transition state where this bond is arranged axially.
Enzymatic Cleavage. A large number of enzymes are known which cleave phosphoester bonds in nucleoside cyclic phosphates. The best known of them is pyrimidyl RNase, the specificity of its action residing in that it hydrolyses the P-O bond formed by the 2'-hydroxyl in pyrimidine nucleoside 2',3'-phosphates:
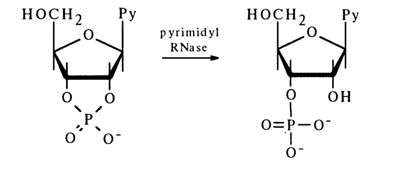
No nucleoside 2'-phosphates are formed in this case. This means that the P-O bond at C3, in the five-membered ring of pyrimidyl RNase is not affected. Purine nucleoside 2',3'-cyclic phosphates are hydrolysed by other enzymes. For instance, guanyl RNase isolated from takadiastase hydrolyses guanosine 2',3'-cyclic phosphates to guanosine 3'-phosphates, with the P-O bond at C3, remaining intact:
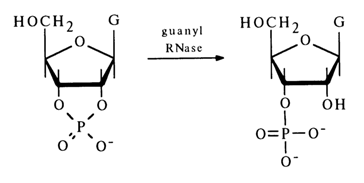
Similar specificity of action is observed in exonuclease A5 isolated from actinomyces.
Phosphodiesterase (PDE) from E. coli hydrolyses nucleoside 2',3'-cyclic phosphates to nucleoside 3'-phosphates irrespective of the nature of the constituent base:
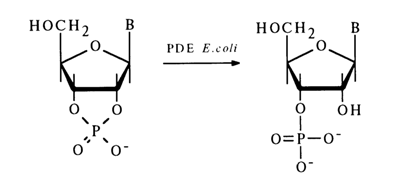
Mammalian tissues have been found to contain PDE hydrolysing nucleoside 3',5'-cyclic phosphates. For example, PDEs isolated from the bovine heart hydrolyse adenosine 3',5'- and uridine 3',5'-eyclic phosphate to the corresponding 5'-phosphates with particular ease.
Other Reactions. In anhydrous alcohols, ribonucleoside 2',3'-cyclic phosphates are rapidly converted, in the presence of hydrogen ions, into a mixture of ribonucleoside 2'- and 3'-alkyl phosphates:
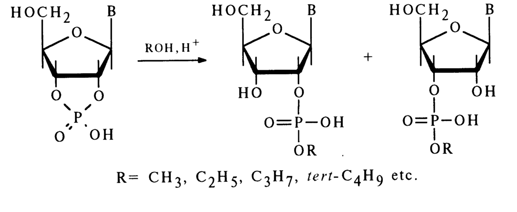
Alcoholates of alkali metals in alcoholic media also hydrolyse ribonucleoside 2',3'-cyclic phosphates to ribonucleoside 2'(3')-alkyl phosphates. This reaction, however, proceeds at a much slower rate, as compared to a similar acid-catalyzed reaction with alcohols, and is accompanied by side processes. For example. treatment of adenosine 2',3'-cyclic phosphate with sodium benzylate in benzyl alcohol results in adenosine 3'- and 2'-benzyl phosphates with a sizable admixture of adenosine 3'- and 2'-phosphates.
The mechanism of these reactions seems to be similar to that proposed for hydrolysis of nucleoside cyclic phosphates, catalysed by acids and bases, with the difference that the nucleophile attacking the tetrahedral phosphorus atom in this case is an alcohol or alkoxy-ion molecule.
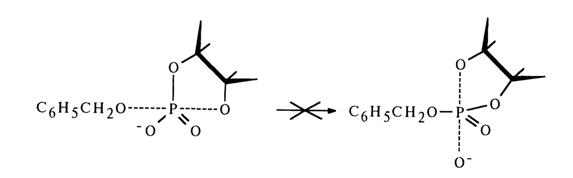
Ribonucleoside 2'- and 3'-alkyl phosphates are easily formed from the corresponding nucleoside 2',3'-cyclic phosphates if a mixed anhydride is first obtained with diphenylphosphoric acid, for example, then treated with excess alcohol. The following conversion mechanism has been proposed:
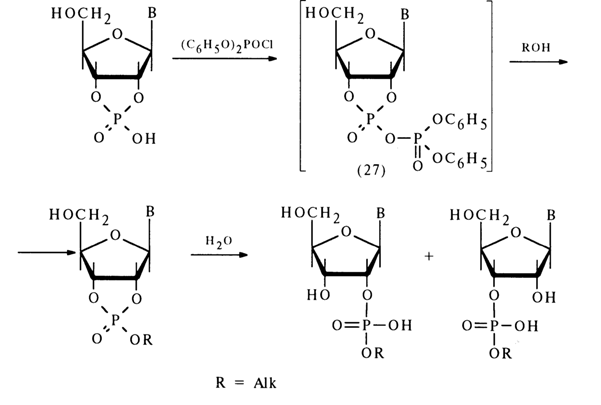
The extremely unstable compound (28) (the lability of its phosphotriester bonds is enhanced by the cyclic nature of the compound) is readily hydrolysed to form nucleoside 2'- and 3-alkyl phosphates. Thus, it is assumed that in the nucleophilic substitution at the phosphorus atom forming part of the five-membered ring of anhydride (27) the leaving group is the diphenylphosphoric acid anion [path (a)]. However, this mechanism lacks corroborating evidence.
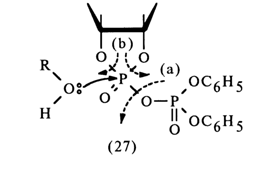
If one proceeds from the general concepts concerning nucleophilic substitution at the tetrahedral phosphorus atom in cyclic phosphates, this mechanism seems unlikely because in this case the O-P-O group of the five-membered ring must occupy an equatorial position in the transition state (axial positions being occupied by the incoming alcohol molecule and leaving diphenyl phosphate group), which will lead to a drastic increase in strain and almost never materializes. Apparently, it is the 2'- or 3'-hydroxyl group of ribose that occupies an axial position and becomes the leaving entity. Thus, when anhydride (27) is attacked by an alcohol (see scheme), two isomeric mixed anhydrides must form:
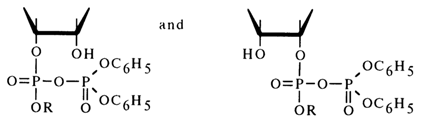
Subsequently, these anhydrides must be easily converted into a cyclic triester (28) as a result of intramolecular nucleophilic substitution at the phosphorus atom. Hence, the conversion of anhydride (27) into cyclic triester (28) seems to involve intermediate anhydrides (29); during this step [(27)
® (28)] the role of the diphenyl phosphate group boils down to enhancing the electrophilicity of the nucleotide phosphorus atom.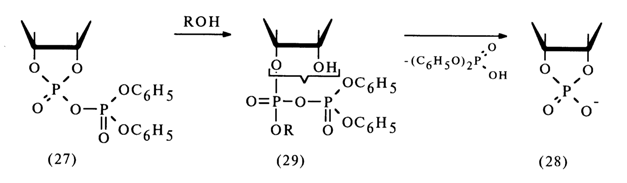
More stable substituted cyclic phosphates are produced from nucleoside 3',5'cyclic phosphates. For example, treatment of uridine 3',5'-cyclic phosphate with diphenyl phosphochloride followed by addition of an amine results in an amide of uridine 3',5'-cyclic phosphate, which has been isolated and characterized:
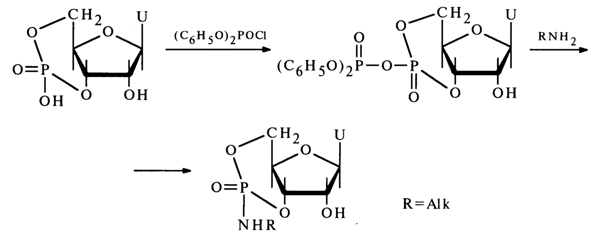
The greater stability of such a trisubstituted cyclic phosphate, as compared to the above-described nucleoside 2',3'alkyl cyclic phosphates, stems from both the greater stability of the six-membered ring and the presence of an amide group at the phosphorus atom.
4.6.2 Alkyl Esters of Nucleotides
The reactions leading to cleavage of phosphoester bonds occupy a special place among the chemical transformations involving alkyl esters of nucleotides. These reactions are widely used in the chemistry of nucleic acids, where they form the basis of the analytical procedures employed to determine composition and structure.
Two principal types of reactions leading to cleavage of the phosphodiester bonds in alkyl esters of nucleotides are known. Those of the first type include hydrolysis catalysed by acids, alkalis and bases and proceeding as nucleophilic substitution at the phosphorus atom with cleavage of the P-O bond. Alkyl esters of nucleotides, with a carbonyl group at the
b-position of the carbohydrate moiety with respect to the alkyl phosphate, are characterized by b-elimination of the latter, accompanied by cleavage the C-O bond. The reactions of both types will be discussed at greater length in what follows.Hydrolysis of Phosphodiester Bonds. Alkyl esters of nucleotides form two distinct groups in terms of phosphodiester bond stability. The first group includes compounds in which the alkyl phosphate group is linked with one of the hydroxyls of the cis-glycol group; that is, ribonucleoside 3'- or 2'-alkyl phosphates (30):
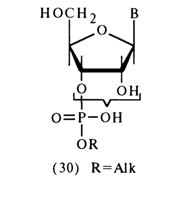
The other group comprises the rest of the alkyl esters of nucleotides; that is ribonucleoside 5'- as well as deoxyribonucleoside 3'- and 5'-alkyl phosphates [(31); X = H in the second compound]:
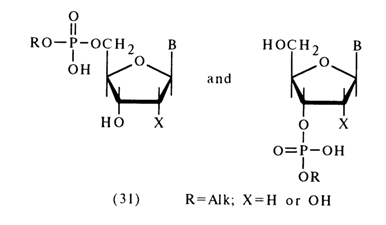
The sharp difference in stability of the phosphodiester bonds in compounds belonging to both groups stems from the presence or absence of a hydroxy group at the
a-position with respect to the phosphate group.Ribonucleoside 3'(2')-alkyl phosphates (30) have a strong resemblance, in terms of their properties, to alkyl phosphoglycols - that is, they are stable only at neutral pH values. Under conditions of alkaline or acid catalysis, these compounds readily dissociate to free nucleotides. Therewith, the cleavage of phosphodiester bonds proceeds, just as in the case of alkyl phosphoglycols, as transphosphorylation, which is to say that the neighboring hydroxyl group of ribose is involved in this instance. The initially formed ribonucleoside 2',3'-cyclic phosphate yields a mixture of nucleoside 3'- and 2'-phosphates during hydrolysis.
The catalytic function of bases B resides in that by forming a hydrogen bond with the hydroxyl group adjacent to the phosphodiester one (with 2'-OH in the scheme below) they render this hydroxyl group more nucleophilic. The result is promotion of the intramolecular nucleophilic attack at the phosphorus atom.
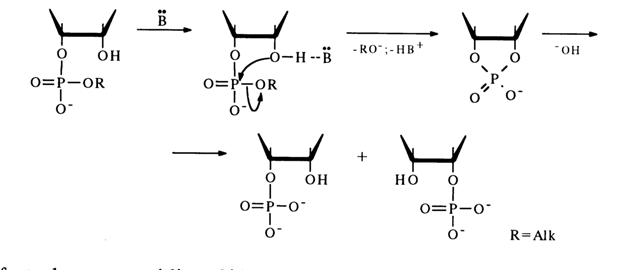
The fact that no cytidine 2'-benzyl phosphate has been found among the products of incomplete alkaline hydrolysis of cytidine 3'-benzyl phosphate suggests that the transition state associated with the nucleophilic substitution at the phosphorus atom, involving the 2'-hydroxyl group, has the structure of a trigonal bipyramid in which axial positions are occupied by the attacking 2'-hydroxyl and leaving benzyl groups. The nucleophilic attack ends in formation of nucleoside 2',3'-cyclic phosphate. In this case, there is no pseudorotation which may result in axial arrangement of, the P-O bond at C3, with subsequent departure of the 3'-hydroxyl group and formation of nucleoside 2'-benzyl phosphate.
After the cyclic phosphate has formed, it undergoes hydrolysis with extreme ease, giving a mixture of nucleoside 2'- and 3'-phosphates without migration of the phosphate group.
During acid hydrolysis of nucleoside 3'(2')-alkyl phosphates (30), migration of the alkyl phosphate group is observed along with cleavage of the phosphodiester bond. For example, during incomplete (by 50 %) acid hydrolysis of cytidine 3'-benzyl phosphate, the reaction mixture was found to contain cytidine 2-benzyl phosphate (25 %) in addition to cytidine 2'- and 3 '-phosphates as well as the unreacted ester.
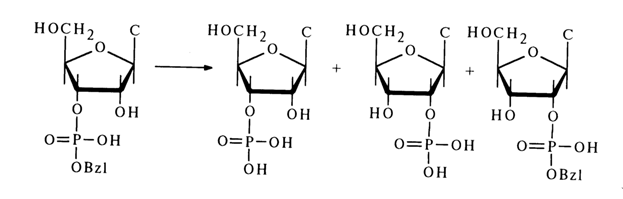
The formation of cytidine 2'-benzyl phosphate may be due to the pseudorotation in the transition state arising in the course of the substitution.
The compounds of structure (31), in which the nucleophilic properties of the neighboring hydroxyl group cannot influence the course of the reaction, behave like dialkyl phosphates or, in other words, are stable toward acid and alkaline hydrolysis. The most stable are such derivatives of adenosine as adenosine 5'- as well as deoxyadenosine 5' - and 3'-benzyl phosphates.
During acid hydrolysis of esters of type (31) under vigorous conditions, the cleavage of phosphodiester bonds is complicated by that of the glycosidic bond (especially in the case of purine nucleotides). The cleavage of phosphodiester bonds, if it is made possible, occurs not only between the oxygen and phosphorus atoms, as was the case with phosphodiesters of structure (30), but also primarily between the oxygen and carbon.
Alkaline hydrolysis of nucleoside phosphate esters of type (31) is facilitated by incorporation of electron-acceptor substituents into the residue R. Interestingly, nucleoside 3',5'-cyclic phosphates may form at the same time, although the transposition of the 3'-hydroxyl and CH2OH groups hampers this reaction. For instance, deoxythymidine 3'-(para-nitrophenyl phosphate) is converted into deoxythymidine 3',5'-cyclic phosphate in an alkaline medium.
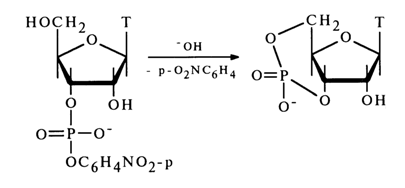
The cyclization (nucleophilic substitution at the phosphorus atom with the participation of a sterically hindered 5-hydroxyl group) is in this case facilitated by the ease of departure of the stable para-nitrophenolate anion.
Two conclusions should be drawn from the finding that ribonucleoside 3'(2')-alkyl phosphates are highly labile. Firstly, the intramolecular nucleophilic attack of the phosphorus atom in nueleoside 3'(2')-phosphates is facilitated by the 2'(3')-hydroxyl group being sterically close to the phosphate group. In the presence of such a hydroxyl group in a nucleotide molecule, the nucleophilic attack of the phosphorus atom by water or a hydroxyl anion proceeds at a slower rate. The intramolecular nucleophilic attack results in an intermediate state with a five-coordinated phosphorus atom, in which one of the cis-hydroxyl groups of ribose (the attacking one) is arranged axially and the other (linked to the phosphate group), equatorially. Another axially arranged substituent is always an alkoxy group. The departure of this group (the main direction of nucleophilic substitution at the phosphorus atom) is followed by formation of a cyclic diphosphate which is easily hydrolysed, unlike dialkyl phosphates, to a mixture of 2'- and 3'-phosphonucleosides. During this step, the attacking group is a water molecule. Thus, if a nucleoside 3'(2')-phosphate contains a 2'(3')-hydroxyl group sterically close to the phosphate group, both emergence of the transition state and the subsequent hydrolysis of the intermediately forming cyclic phosphate are accelerated. Consequently, the neighboring hydroxyl group catalyses the hydrolysis of nucleoside 3'(2')-alkyl phosphates (a similar pattern is observed in the case of polyol phosphates as well).
The other important aspect of hydrolysis of ribonucleoside 3'(2)-alkyl phosphates is the fact that it involves different mechanisms depending on whether the medium is acid and alkaline.
If the process is catalysed by an acid, the intermediate state (32) arising from ribonucleoside 3'-alkyl phosphate undergoes two transformations: conversion into cyclic phosphate (33) and pseudorotation as a result of which the nondissociated hydroxyl of the phosphate group and that of ribose, associated with the alkyl phosphate proup prior to the reaction [structure (36)l occupy axial positions. If the hydroxyl group of ribose "leaves" such a state, migration of the alkyl phosphate group takes place and the emerging isomer (34) again undergoes the same transformations involving the hydroxyl group adjacent to the phosphodiester bond and the end products are the nucleoside 2'- and 3'-phosphates forming in equal amounts.
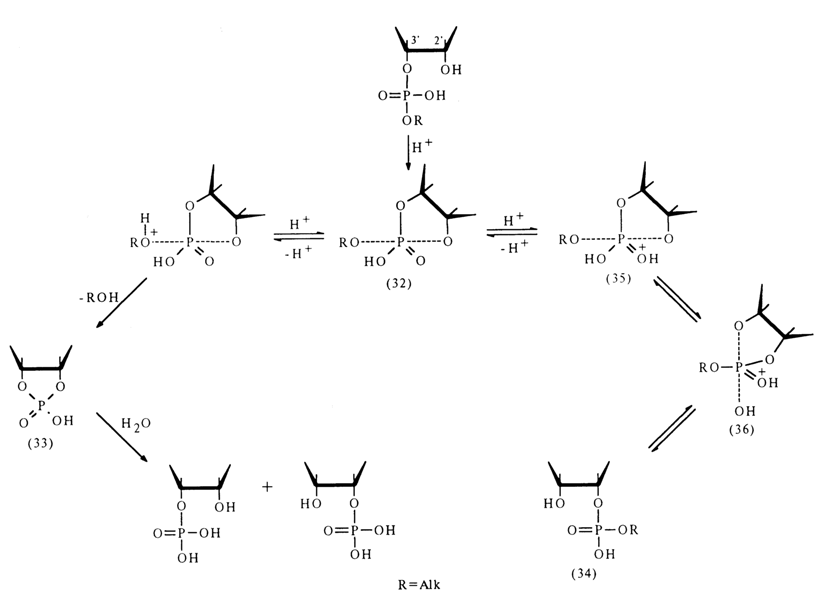
Pseudorotation in intermediate state (32) is impossible because of the absence of two sufficiently electronegative groups in the equatorial position, which could "stretch" the equatorial P-O bonds to the length of axial ones. After protonation. structure (32) may undergo conversion into structure (35) where such groups are already present (hydroxyl and 3'-O-CH< group). Therefore, pseudorotation and subsequent transition toward compound (34) become possible.
During alkaline hydrolysis, the P-O bonds cannot be equalized in length with subsequent pseudorotation in transition state (32) because the bonds linking the phosphoryl oxygen atom and that of the oxygen resulting from the dissociation (P-OH
® P-O-) to the central phosphorus atom remain much stronger than the rest of the P-O bonds.4.6 Properties of Nucleotide Derivatives ... 157
This is why the transformations catalysed by bases are not accompanied by migration of the alkyl phosphate group. The products of alkaline hydrolysis of nucleoside 3'(2')-alkyl phosphates at any step of the process are nucleoside 3'- and 2'-phosphates.
Hydrolysis of Phosphotriester Bonds. Triesters, which are analogues of diesters of types (30) and (31), also differ in stability of the phosphotriester bonds.
Triesters in which the nucleophilic behavior of the neighboring hydroxyl cannot affect the course of the reaction are in many ways similar to trialkyl phosphates.
As regards stability of phosphotriester bonds, compounds contanining a hydroxyl next to the phosphotriester group stand distinctly apart:
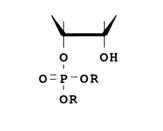
Such triesters should behave like protonated diesters. They correspond to an activated form (30, H = R) that initiates diester cleavage and are relatively unstable. Assuming an S,,2 mechanism, one can expect formation of a cyclic triester as an intermediate product after the elimination of an OR anion. This intermediate will be hydrolysed by a water molecule to a mixture of 2'- and 3'-phosphodiesters:
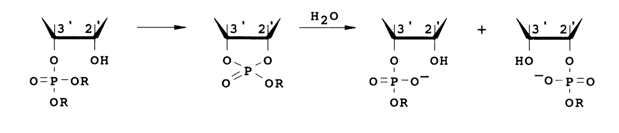
Since only the starting triester and 3'(2')-diesters can be observed during the reaction, the rate-limiting step must be either attack of the OH group or cleavage of the ester bond. The reaction rates are pH-dependent. At pH ranging from 4.5 to 8.5, they increase by about a factor of two per pH unit during hydrolysis of 2'- and 3-triesters. This dependence of the reaction rates on pH can be ascribed to the following factors: increasing nucleophilicity of the attacking OH group and decreasing electrophilicity of the phosphorus (both factors influencing the rate of the attack).
Enzymatic Cleavage. Some of the enzymes hydrolysing phosphodiester bonds in nucleoside cyclic phosphates also break down noncyclic esters of nucleotides. A case in point is pyrimidyl RNase which catalyses the hydrolysis of pyrimidine nucleoside 3'-alkyl phosphates, 2-ketopyrimidine derivatives without any substituent at N3. The result is the corresponding nucleoside 3'-phosphates or their derivatives. Nucleoside 2',3'-cyclic phosphates emerge as intermediates. For example:
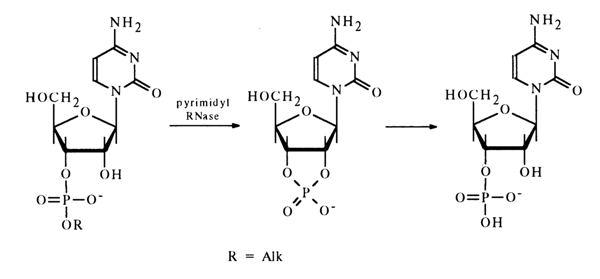
Pyrimidyl RNase cleaves the coresponding phosphodiester bonds formed by uridine and cytidine derivatives, as well as some minor components of RNA, such as pseudouridine, 5,6-dihydrouridine, and ribothymidine.
The enzyme does not hydrolyse pyrimidine ribonucleoside 2'- and 5'-alkyl phosphates, nor does it break down any purine ribonucleoside alkyl phosphates. Pyrimidine and purine deoxyribonucleoside 3'(5')-alkyl phosphates are not hydrolysed by pyrimidyl RNase either.
This enzyme with its capacity for breaking phosphodiester bonds in pyrimidine ribonucleotides only with a particular position of the alkyl phosphate group is widely used in structural studies because it permits one to determine the structure of nucleoside alkyl phosphates as well as enables controlled hydrolysis of ribonucleic acids.
Guanyl RNase (derived from takadiastase or actinomyces 9)) catalyses the hydrolysis of ribonueleoside 3'-phosphates, derived from 6-ketopurines unsubstituted at Nl and N7 They include guanosine among the major RNA components and inosine, N2 -methyl guanosine and N2 -dimethyl guanosine among the minor ones (see Table 1-5). Just as in the above-described cases, this reaction involves a cyclic phosphate formation step, for example.
9)
These two preparations of guanyl RNAse differ in that the activity of one of them (isolated from actinomyces) is less dependent on the presence of substituent at Nl and N7 in the guanine nucleus of the subastrate.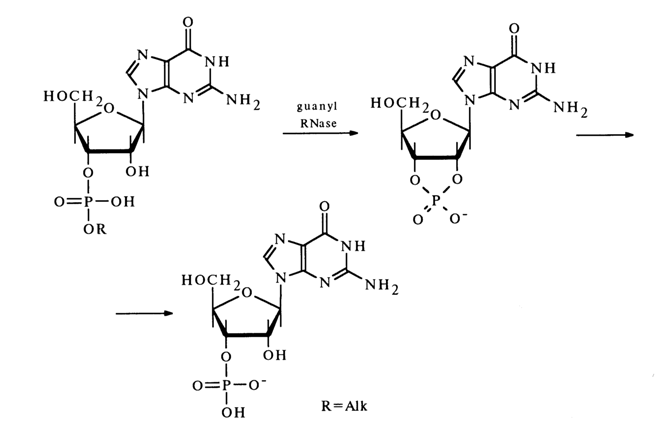
In some instances, the second step of the reaction, namely, hydrolysis of nucleoside 2',3'-cyclic phosphates, proceeds at a very slow rate so that the latter accumulate in sizable amounts. Guanyl RNase does not catalyze the hydrolysis of alkyl phosphates of other ribo- and deoxyribonucleosides.
By virtue of its highly specific action, guanyl RNase is the major enzyme used in determining the RNA structure.
In addition to the nueleases considered above, ther is a number of enzymes that are less specific. Worthy of mention among the enzymes most frequently used for breaking phosphodiester bonds are phosphodiesterases (PDEs) isolated from the spleen and micrococci, which catalyze the breaking of virtually any nucleoside 3'-alkyl phosphates down to the corresponding nucleoside 3'-phosphates. Another commonly used enzyme is PDE from snake venom, which, in contrast to the PDEs mentioned above, catalyses the hydrolysis of nucleoside 5'-alkyl phosphates to the corresponding nucleoside 5'-phosphates.
b
-Elimination reactions. The cleavage of phosphodiester bonds in alkyl esters of nucleotides may be accompanied by breaking not only of P-O but also C-O bonds. The latter must, in this case, be activated by the b-carbonyl group in pentose. The incorporation of such a carbonyl group may be achieved either through oxidation of the hydroxyl groups in the carbohydrate moiety, which leads to activation of the 5'- and 3'-phosphoester bonds, or through elimination of the heterocyclic base, which leads to activation of the 3'-phosphoester bond.Notably, the products of periodate oxidation of nucleoside 5'-alkyl phosphates lend themselves readily to hydrolysis in an alkaline medium. This entails cleavage of the C-O bond with detachment of the phosphomonoester:
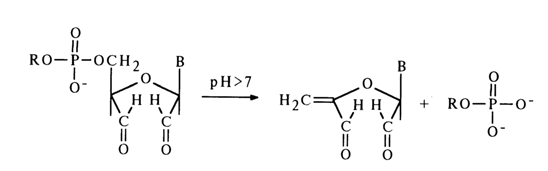
Along with
b-elimination of the alkyl phosphate group, the oxidized nucleoside in an alkaline medium often undergoes cleavage of the N-glycosidic bond with separation of the heterocyclic base. Primary amines serve as effective catalysts of such cleavage.The 5'-alkyl phosphate group may also be eliminated in the corresponding deoxynucleotide derivatives after oxidation of their secondary hydroxyl group. For instance, when solutions of deoxynucleoside 5'-alkyl phosphates are heated in a mixture of acetic anhydride and pyridine with dimethyl sulfoxide, deoxyribose is oxidized with subsequent P-elimination of the alkyl phosphate group and simultaneous cleavage of the N-glycosidic bond:
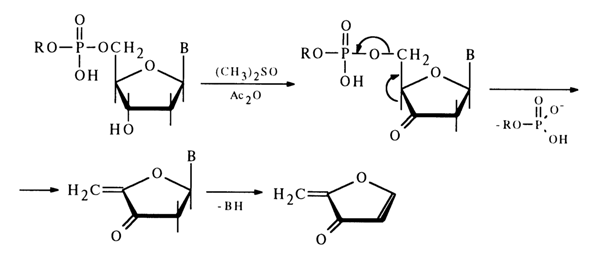
In the case of deoxynucleoside 3'-alkyl phosphates, a similar cleavage of the phosphodiester bond can be easily achieved in an alkaline medium after oxidation of the primary hydroxyl group with atmospheric oxygen over a platinum catalyst but with preliminary conversion of the carboxyl into an amide group:
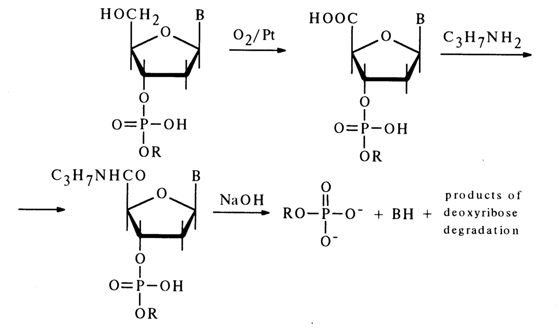
4.6.3 Mixed Anhydrides of Nucleotides
As has already been mentioned, mixed anhydrides of nucleotides occur widely in nature and play a major role in many biosynthetic processes. The most important of them are phosphoric acid derivatives known as nucleotide coenzymes or nucleotide anhydrides 10).
Anhydrides with Phosphoric Acids. It has already been mentioned that anhydrides of nucleotides and diphenylphosphoric acid are highly reactive, their instability making it impossible to isolate these compounds, but they can be made to react with various nucleophilic agents in an absolute solvent. In such cases, the nucleophile replaces the diphenylphosphate group to yield the corresponding nucleotide derivatives. Anhydrides of nucleotides with monoesters of phosphoric or unsubstituted phosphoric and pyrophosphoric acids are less reactive. Such anhydrides are stable in neutral media but are hydrolysed in acid ones. At alkaline pH values, they exist in the form of di- or trianions, which is why it is difficult for the hydroxyl anion to approach them because of electric repulsion. As a result, they are relatively stable toward alkaline hydrolysis. Cleavage of the pyrophosphate bond is easy in an alkaline medium as well if the pyrophosphate group is next to a hydroxyl one; in this case, the reaction is accompanied by formation of a cyclic phosphate. For instance, treatment of cytidine 5-diphosphoglycerol with an aqueous solution of ammonia gives cytidine 5'-phosphate and glycero-1,2-cyclic phosphate:
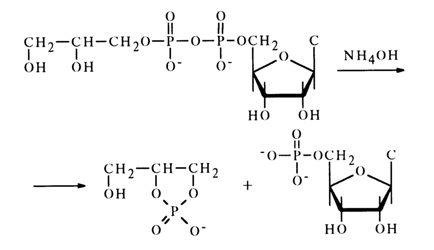
Anhydride of cytidine 5'-phosphate with phosphoric or pyrophosphoric acid is stable under the same conditions. The role of the neighboring hydroxyl in such anhydrides is reminiscent of the involvement of 2'(3)-hydroxyl of ribose in alkaline hydrolysis of ribonucleoside 3'(2')-alkyl phosphates.
The effect of the hydroxyl group that may lead to formation of a six-membered cyclic phosphate is less pronounced. For example, coenzyme A (37) partially dissociates to a cyclic phosphate when treated with an alkali.
10)
The chemistry of these compounds is treated in the well known monograph by A.M. Michelson (see references at the end of this chapter).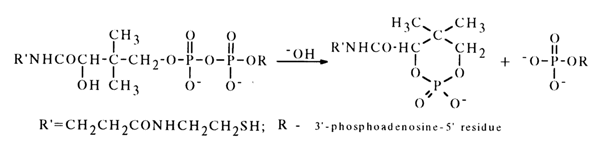
When anhydride of deoxythymidine 5-phosphate is heated in pyridine, the pyrophosphate undergoes partial conversion into deoxythymidine 3',5'-cyclic phosphate:
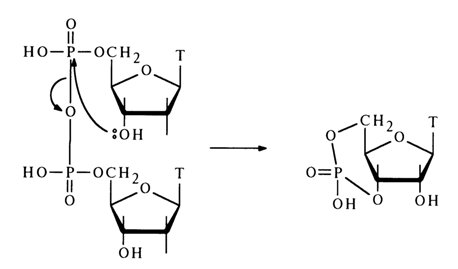
Descriptions of intermolecular reactions between such anhydrides and nucleophilic agents are scant. Only a pyrophosphorolysis reaction involving nucleotide anhydrides is more or less well known. For example, anhydride of adenosine 5'-phosphate reacts with pyrophosphoric acid in pyridine to yield adenosine 5'-triphosphate:
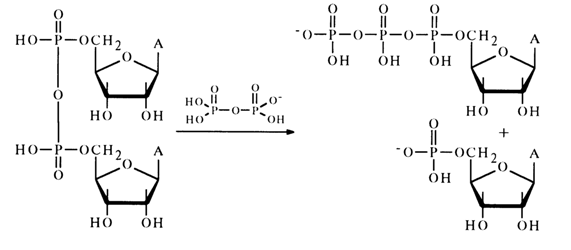
Moreover, the reaction mixture was found to contain adenosine 5'-diphosphate which seems to result from nucleophilic substitution for the terminal phosphate group in the triphosphate:
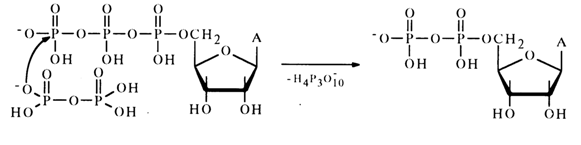
Anhydrides with Carboxylic Acids. Mixed anhydrides of nucleotides and carboxylic acids belong to the most important biologically active nucleotide derivatives. Formation of a mixed anhydride of adenosine 5'-phosphate and an amino acid marks the beginning of activation of the latter, necessary for its incorporation into the peptide chain during protein biosynthesis in ribosomes. This reaction, proceeding in the presence of the enzyme aminoacylTRNA ligase specific for each amino acid, resides in nucleophilic attack of the a-phosphate link in adenosine 5'-triphosphate by a free amino acid.
A similar activation mechanism has been established for acetic, butyric and other carboxylic acids participating in the biosynthesis of steroids and other biologically important compounds.
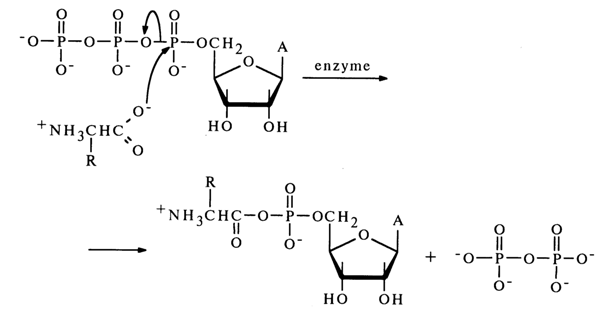
Before discussing the properties of mixed anhydrides of nucleotides with carboxylic acids, we must take a brief look at those of their simpler analogues acyl phosphates.
General Concepts About Acyl Phosphates. Acyl phosphates feature two reactive sites that can be attacked by nucleophiles - phosphorus in the phosphate group and carbon in the carbonyl.
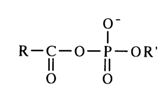
However, due to differences in the structure of carboxyl and phosphate groups, the mechanisms of nucleophilic substitution at the above-mentioned atoms are widely dissimilar. The effectiveness of the attack at the carbonyl carbon is determined by the ease of linkage with the nucleophile, whereas the decisive factor for the nucleophilic attack at the phosphate phosphorus is the nature of bonding with the group being substituted (the course of both reactions is also materially affected by the nature of the substituents, nucleophilic agent and solvent). In order to demonstrate the peculiar behavior of acyl phosphates in nucleophilic substitution reactions, it would be appropriate at this juncture to compare the behavior of asymmetric mixed anhydrides of two carboxylic acids and asymmetric pyrophosphates.
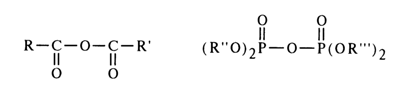
The nucleophilic agent Nu attacks the mixed anhydrides of carboxylic acids at the most electrophilic site - that is, at the carbonyl carbon of a stronger acid (RCOOH). The result is the corresponding derivative of the latter, and the function of the leaving group is performed by the anion of the weaker acid (R'COO-).
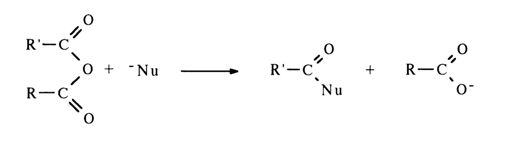
The mixed anhydride of two phosphoric acids, or asymmetric pyrophosphate, is attacked by the nucleophile at both phosphorus atoms; the effectiveness of the attack in this case, however, is determined not by the magnitude of the partially positive charge at these atoms but by the nature of the leaving group: the more stable the departing anion (i. e., the stronger the acid corresponding to the anion), the easier the nucleophilic substitution. Thus, if the first of the two phosphoric acids (R"O)2P(O)OH and (R'''O)2P(O)OH corresponding to the asymmetric pyrophosphate shown above is stronger, the attack at the phosphorus of the second acid will be more effective:
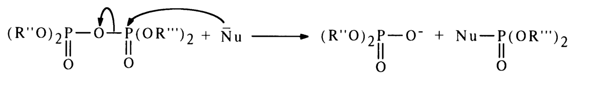
Consequently, the nucleophile receives the residue of the weaker phosphoric acid.
Acyl phosphates are most likely to exhibit properties of both carboxylic anhydrides and pyrophosphates. Hence, it becomes clear that they can serve as both acylating and phosphorylating agents. The path of the reaction is determined by many factors, including the nature of the nucleophilic agent, the degree of screening of the reactive sites (carbonyl carbon and phosphorus of the phosphate group), the type of solvent, the thermodynamic stability of the reaction products, and some others. More often than not, reactions between acyl phosphates and nucleophilic agents in the absence of enzymes result in transfer of the acyl group to the nucleophile (yielding the corresponding carboxylic acid derivative).
Acyl phosphates of the general formula
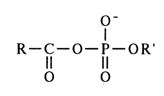
act as acylating agents, provided the carboxylic acid is stronger than the correspoding phosphoric one (pK of the secondary hydroxyl of the phosphate group, involved in anhydride bonding, is in the neighborhood of 6.0, whereas pK of the carboxyl group in carboxylic acids usually ranges from 2 to 4). The path of the reaction seems to be determined by the greater electrophilicity of the carboxyl carbon and its accessibility as a result of two-dimensional structure of the carboxyl group.
The interaction of acyl phosphates with amines usually leads to acylation of the latter. The following reaction between acyl phosphates and hydroxylamine is especially well known:
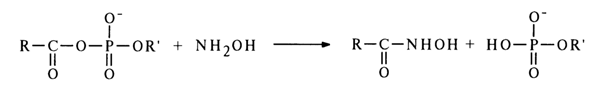
The reaction proceeds with extreme ease. At pH 5-7, for example, the quantitative formation of hydroxamic acid takes ten minutes; the latter gives colored complexes with iron salts, which are useful in quantitative estimation of the acyl phosphate content in the reaction mixture. The hydroxamic reaction is known to be the standard of reactivity of carboxylic acid derivatives.
The interaction of acetyl phosphate with tertiary amines (as opposed to primary and secondary ones) can lead not only to acetylation but also phosphorylation of the amine:
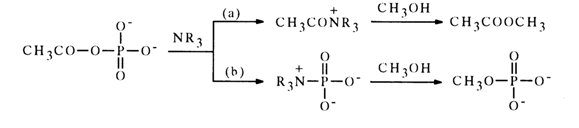
Depending on the structure of the tertiary amine, the reaction may proceed predominantly along each of the indicated paths (a) and (b). For instance, the acylation of imidazole and trimethylamine proceeds along either path with equal probability, while in the case of pyridine phosphorylation is the predominant path [path (b)].
In contrast to acetyl phosphate, the reactions of acyl phosphates with primary, secondary and tertiary amines yield acylated amines:
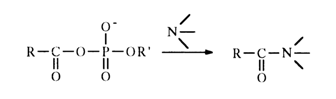
The rates of such reactions are usually an order of magnitude higher than in the case of simple acetyl phosphate.
The mild conditions of such reactions and the high yields of acylated amines form the basis of one of the peptide synthesis methods:
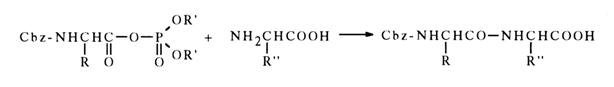
When the nucleophilic attack of the carbonyl carbon in an acyl phosphate is hindered, phosphorylation becomes the predominant reaction path, for example:
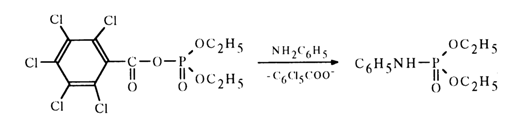
Let us now consider acyl phosphates of more complex structure, in which the function of the phosphate moiety is performed by nucleotides.
Anhydrides of Nucleotides and Carboxylic Acids. As has already been mentioned, such anhydrides can be synthesized and isolated individually. Some nucleotide anhydrides belonging to the class of acyl phosphates may be stored for long periods of time. Among the most labile nucleotide derivatives of this type are anhydrides with acetic and other fatty acids as well as with amino acids.
These compounds are easily hydrolysed as a rule and react with hydroxylamine and amines. All these reactions lead to transfer of the acyl group to the nucleophile. The reaction with hydroxylamine (hydroxamic reaction) is used, just as in the case of simple acyl phosphates (see above), for quantitative determination of such anhydrides. For instance:
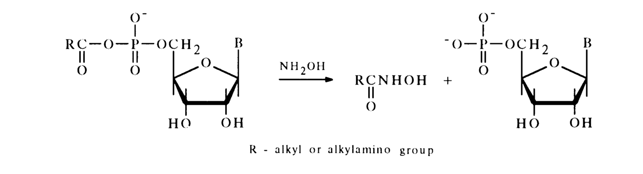
The reaction with phosphoric acid in pyridine proceeds in a similar manner. For example:
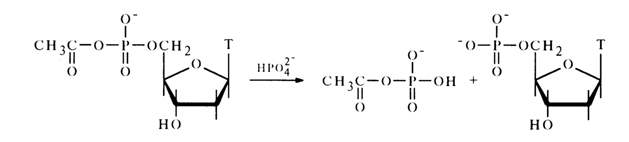
The resulting acetyl phosphate reacts further with the next phosphoric acid molecule to yield an inorganic pyrophosphate:
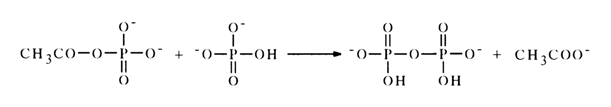
The reverse reaction in pyridine is virtually nil, and the equilibrium shifts toward formation of pyro- and polyphosphates. The partial conversion of nucleotides into symmetric pyrophosphates is usually observed when they are exposed to carboxylic anhydrides in order to incorporate protective acyl groups (at the hydroxyl groups of pentose or into the nucleus of the heterocycle).
Special properties are displayed by anhydrides of ribonucleoside 3'(2')-phosphates and carboxylic acids, which are easily converted, due to proximity of the 2'(3')-hydroxyl group of ribose, into the corresponding nucleoside 2',3'cyclic phosphates. Thus, unlike anhydrides of ribonucleoside 5'-phosphates with carboxylic acids, their 3'-isomers are essentially phosphorylating rather than acylating agents. A similar change in properties from 5'- to 3'-derivatives seems to be caused by that the phosphate group in the latter is sterically close to the hydroxyl one, which results in some kind of intramolecular catalysis. Consequently, it may be assumed that anhydrides of nucleoside 5'-phosphates with carboxylic acids can act as phosphorylating agents if they are formed at intermediate steps of biosynthetic processes or if an enzyme ensures approach of the nucleophilic agent to their phosphate, and not carboxyl, group.
There is no reason why the possibility of such reactions should be ruled out, although they are yet to be observed.
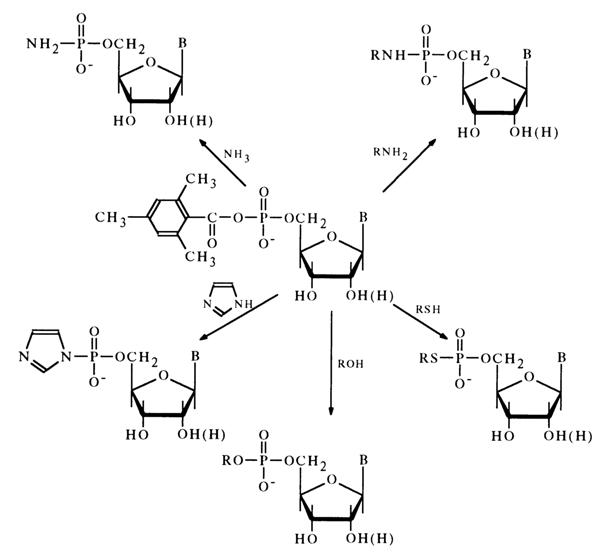
The most stable of the known anhydrides of nucleotides and carboxylic acids are mesitylenecarboxylic acid derivatives. They can be isolated individually and stored for a long period ot time. Studies into the reactions of these anhydrides with nucleophilic agents have shown that their acylating function is inhibited. In reactions with nucleophilic agents, anhydrides of nucleotides and mesitylenecarboxylic acid behave like phosphorylating agents.
If, however, the reaction with amines proceeds rather easily in aqueous solutions, in the case of weaker nucleophiles, such as mercaptans and alcohols, better results are obtained in organic solvents.
Thus, mixed anhydrides of nucleotides with mesitylenecarboxylic acid were the first representatives of "active" nucleotide derivatives that could be isolated individually and used in further reactions. What makes this class of acyl nucleotides promising is that nucleotide or polynucleotides can be linked covalently with their aid to the nucleophilic sites of various substances, the appropriate reaction being conducted in aqueous media (anhydrides of nucleotides with mesitylenecarboxylic acid are not hydrolysed at neutral pH values and at room temperature). It may be assumed, in particular, that these anhydrides can be used as inhibitors of nucleotide metabolism enzymes at least when the active site of the enzyme is occupied by a sufficiently active nucleophilic group (imidazoles, c-amino group of lysine, SH group).
4.6.4 Amides of Nucleotides
Amides of nucleotides - derivatives of aliphatic and aromatic amines - usually are rather stable. The presence of an amide moiety in the nucleotide molecule naturally gives rise to some specific properties standing distinctly apart from those of the corresponding alkyl esters. In view of the important role of amides of nucleotides in synthesis, along with relatively scant information about them, discussed in what follows are simple amidophosphates whose properties are in many ways similar to those of nucleotide amides.
General Concepts About Amidophosphates. The reactivity of amidophosphates is strongly dependent on the form in which they react: that of a conjugated acid (38), neutral molecule (39), or anion (40):
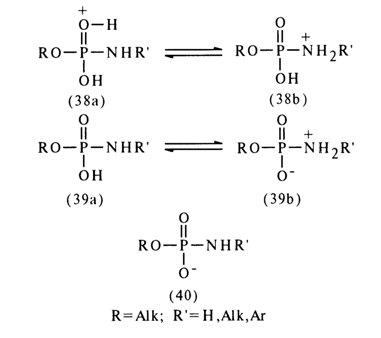
The conjugated acid (38) seems to represent a mixture of two forms differing in the site of proton attachment: form (38a) is marked by protonation of the phosphoryl oxygen, while form (38b) is marked by that of the amide nitrogen. The proton in the neutral form (39) may also occur either at the oxygen (39a) or at the nitrogen (39b). The anionic form of the amidomonophosphate carries a negative charge concentrated primarily at the oxygen atom.
The hydrolysis rate of amidophosphates is to a great extent dependent on the pH of the medium and structure of the amide moiety. Aliphatic amine derivatives are highly unstable in acid media (the hydrolysis rate in this case varying with the acid concentration), hydrolysed slowly at pH 4-6 and virtually stable at alkaline pH values. Hence, the conjugated acid (38b) is the most reactive form. It is quite possible that hydrolysis of the neutral form is determined by the presence of molecules protonated at the nitrogen (39b). Amidophosphates that are derivatives of aromatic amines are hydrolysed in acid media at a slower rate than aliphatic amine derivatives, which seems to stem from the difficulties in protonation of the amide nitrogen whose lonepair electrons are involved in conjugation with the aromatic nucleus. The relationship between the hydrolysis rate and capacity of the amide nitrogen for protonation is supported by the results of hydrolysis of N-acyl amides.
Such compounds are hydrolysed more easily at pH 4, whereas at lower and higher pH values the hydrolysis rate drops sharply, which is strongly reminiscent of the behavior of monophosphates. In this connection, the following hydrolysis mechanism (through an intermediate hydration step) has been proposed for them:
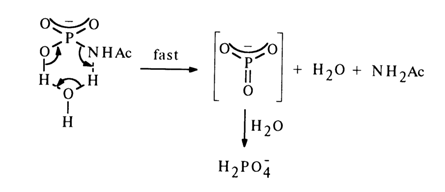
Proceeding from the above data and similar evidence concerning the behavior of unsubstituted amidophosphate, the following mechanisms of hydrolysis and solvolysis of amidophosphates have been proposed.
The bimolecular (associative) mechanism is similar to the
SN2 mechanism of nucleophilic substitution for other phosphoric acid derivatives containing a suitable leaving group. In accordance with this mechanism, the stability of the monoanionic form (40) toward nucleophilic substitution is due to the strong pp-dp conjugation of the phosphorus and nitrogen atoms, which leads to occupation of the 3d orbitals of the phosphorus and hinders the formation of an intermediate compound of the trigonal bipyramid type during the nucleophilic attack. Moreover, departure of the amide group in such a form is disadvantageous. In the case of the protonated form (39b), the situation is altogether different because the 3d orbitals become vacant as a result of the upset pp-dp conjugation and the protonated amide group is apparently ready to "depart" in the form of amine. Still more reactive for the same reason is the form (38b) of conjugated acid, as the conjugation of the phosphorus and oxygen atoms is less pronounced because of no negative charge being present at the latter.As regards hydrolysis and solvolysis of the neutral form of amidomonophosphates, the bimolecular mechanism is most likely. For example, methylphosphate cyclohexylamide is converted into methylethyl phosphate by less than ten per cent during solvolysis in 50 % ethanol.
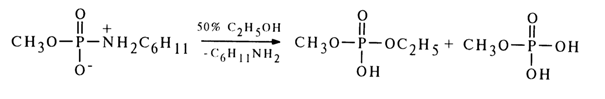
If the nucleophilic substitution for this amide had been based on the SN1 type mechanism (if only the substrate molecule were involved in the rate-determining step), no selectivity with respect to one of the nucleophilic agents would have been observed because the rates of the reaction with a molecule of water and alcohol would have been the same.
The monomolecular (dissociative) mechanism is analogous to the SN1 mechanism of nucleophilic substitution at the phosphorus atom, which boils down to elimination of the labile metaphosphate, promoted, just as in the case of the SN2 mechanism, by protonation of the amide nitrogen:
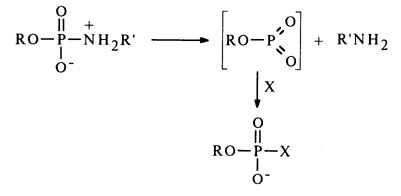
The distinguishing feature of this mechanism, as opposed to the bimolecular one, is lack of selectivity toward the nucleophile.
Such a mechanism has been proposed for the above-described reaction of solvolysis of methylphosphate cyclohexylamide in 50% ethanol, but in the presence of pyridine, since the yield of methylethyl phosphate in this case practically corresponds to the ethanol content in the reaction mixture.
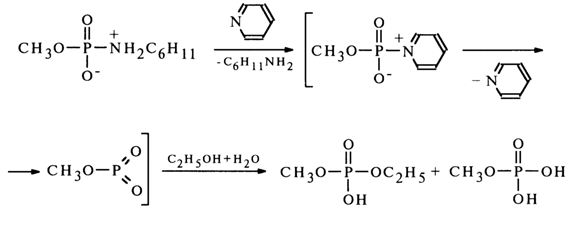
The intermediate compound in this case seems to be methylphosphorylpyridinium which is then converted into methylmetaphosphate. A monomolecular mechanism has also been proposed for solvolysis of the monoanionic form of N-arylamidophosphates.
Intermediate Mechanism (1a). Some reactions are believed to involve a transition state intermediate between trigonal bipyramid and planar metaphosphate. Such a transition state is characterized by a lengthening of the P-N bond and a shortening of the bond which links the phosphorus atom with the incoming nucleophile. Transformations of this kind possess features of both mono- and bimolecular reactions (as regards the effect produced by the nucleophile). A corresponding transition state is believed to be involved in solvolysis of the monoanionic form of the unsubstituted amidophosphate (40).
This hypothesis is based on the fact that hydrolysis of the amidophosphate is 103
times faster, as compared to that of para-nitrophenyl phosphate, although if the reaction with water were based on an SN2 type mechanism, the unsubstituted amidophosphate would be expected to react at a slower rate than the aromatic ester. On the other hand, if the reaction of solvolysis in aqueous alcohol proceeded through formation of a metaphosphate, then alkyl phosphate and phosphoric acid would be yielded in the same ratios as during solvolysis of para-nitrophenyl phosphate, whose monomolecular mechanism is universally accepted. However, the amidophosphate yields twice as much alkyl phosphate as para-nitrophenyl phosphate. These findings suggest both mono- and bimolecular mechanisms of solvolysis of the unsubstituted amidophosphate monoanion.In addition to hydrolysis and alcoholysis, amidophosphates also enter into other reactions of nucleophilic substitution for the amino group. Most typical in this respect are reactions with such nucleophiles as phosphoric acid and its esters, carboxylic acids, and amines. During hydrolysis of benzyl amidophosphate, the anion of benzylphosphoric acid acts as an amino group substituting nucleophile. In the presence of two moles of water, this reaction yields an ammonium salt of symmetrical dibenzyl pyrophosphate:
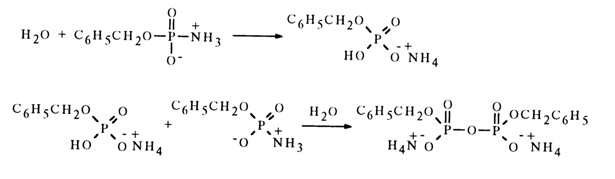
In the absence of water, monoamide of dibenzyl pyrophosphate is formed in inert solvents. In this case, an amidophosphate molecule protonates the amino group of the other molecule to give a nucleophile (amidophosphate anion) and a substrate containing a sufficiently electrophilic phosphorus atom and an appropriate leaving group (amido-phosphate protonated at the nitrogen atom):
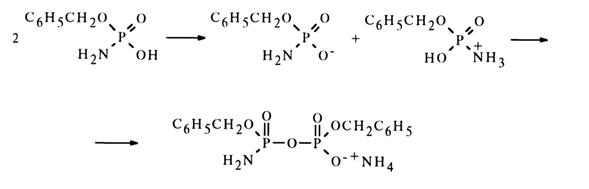
This reaction was studied on many substances and now underlies the preparative method of synthesis of nucleoside 5'-pyrophosphates and related compounds.
Amidomonophosphates also react with carboxylic acids to yield mixed anhydrides. For instance, benzyl amidophosphate dissolved in glacial acetic acid gives acetylbenzyl phosphate. Apparently, in this case, too, protonation of benzytamidophosphate takes place during the first step (see equation above). The protonated form of amidophosphate (at the nitrogen atom) is then nucleophilically attacked not by the like anion, as in the above-described case, but by excess acetic acid.
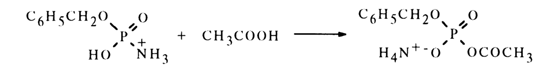
Similarly, the function of nucleophiles can be performed by amines. In this case, one amino group at the phosphorus atom is replaced by another (such reactions are referred to as transamination). Just as the above-considered reactions of substitution of the amino group in alkylamidomonophosphates, transamination seems to involve the protonated form of amidophosphates. A case in point is reactions involving methylphosphate cyclohexylamide.
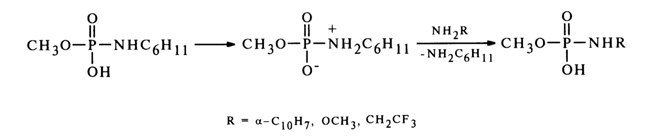
Notably, used as nucleophiles in this case are sufficiently weak amines which do not produce stable ammonium forms in the course of the reaction and. therefore, retain their nucleophilic properties.
The most reactive amides of phosphoric acid and its esters are imidazolides. Imidazolides of mono- and dialkyl phosphates react with phosphoric acid and its esters to give pyrophosphates with a quantitative yield. They also react readily with carboxylic acids, amines and alcohols:
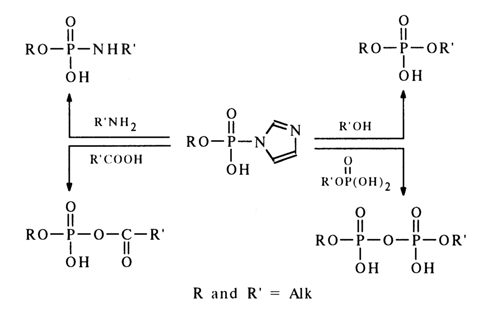
The high reactivity of phosphoimidazoles with respect to nucleophiles is most likely to stem from the ease of intramolecular protonation of the second nitrogen in the imidazole, resulting in an active form in which the phosphorus is linked with the positively charged nitrogen:
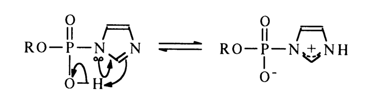
The ease of formation of an active ammonium form in the case of imidazolides seems to be due to the protonation of the nitrogen not linked with the phosphorus and having its basic properties unaffected by the proximity of the phosphate group.
The evidence discussed in this section suggests that amidomonophosphates are stable and rather reactive compounds.
Amides of Nucleotides. Amides of nucleotides are essentially amidomonophosphates as well. It would be natural to assume that many properties of these compounds are more or less very similar. As will be shown here, this is true in some respects, and the many regularities established for amidophosphates also apply to the corresponding nucleotide derivatives. It is for this reason that comparison of the reactivities of these two classes of compounds makes it possible to put knowledge about the properties of the most thoroughly studied simple derivatives to practical use in the chemistry of nucleotide amides.
Insofar as their stability toward hydrolysis is concerned, nucleotide amides resemble closely simple amides of alkyl phosphates: they are stable in aqueous solutions at neutral and alkaline pH values and lend themselves readily to hydrolysis in acid media. As can be inferred from comparative data on the hydrolysis rate of amide derivatives of uridine 5'-phosphate, the rate of the reaction (0.05 N HCI, 370C)
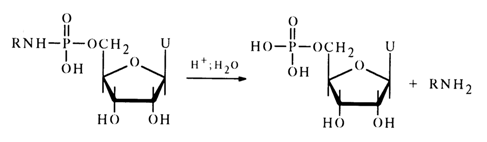
depends on the corresponding amine species, the relationship between the two being rather complex:
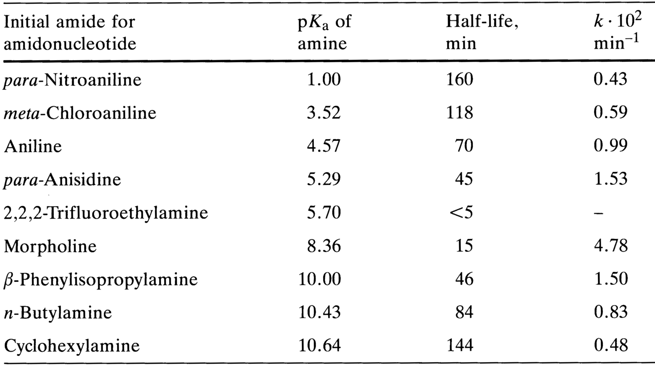
The highest stability toward hydrolysis is displayed by amides - amine derivatives exhibiting weak as well as strong basic properties. Since, as has already been mentioned, hydrolysis of amidophosphates, similarly to reactions of nueleophilic amino group (or substituted amino group) substitution, begins with protonation of the amide nitrogen, it is natural to assume that the rate of the process will depend precisely on this reaction step. This assumption may explain the above regularity in view of the fact that in both extreme cases (when the corresponding amine is either a very weak or a very strong base) the amide nitrogen becomes electron-deficient due to the electron-acceptor influence of the radical at the nitrogen, in the case of amides derived from amines with extremely weak basic properties, or due to the substantial increase in the pp-dp conjugation between the nitrogen and phosphorus atoms, in the case of amides containing strong electron-donor substituents at the same position (which are, consequently, derivatives of a strong base).
Amidonucleotides, just as simple amidophosphates, react rather easily with phosphate anions. Uridine 5'-amidophosphate, for example, yields the corresponding pyrophosphate while reacting with phosphoric acid and its esters in pyridine.
The reactions apparently involve the same mechanism as in the case of alkyl amidophosphates (see above).
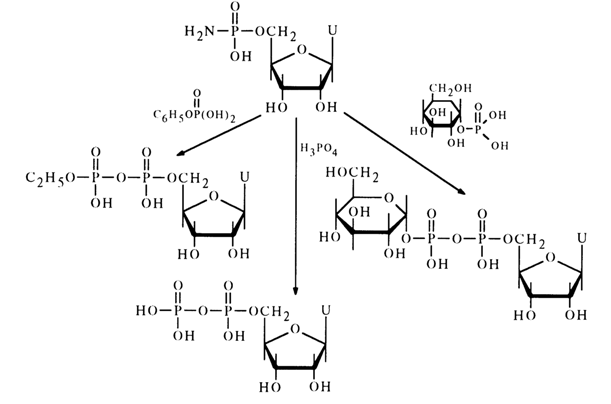
Various amidonucleotides differing in basicity of the starting amine have been used in reactions with phosphate anions: anisidides, morpholides, piperidides, and cyclohexylamides. Nucleotide morpholides are most commonly used in the synthesis of nucleoside 5'-pyrophosphates and analogous compounds. One of the reasons for the reactivity of morpholides, just as in the case of acid hydrolysis, may be the ease of emergence of the activated protonated form. Reactions of morpholides of uridine 5'-, guanosine 5'-, adenosine 5'- and cytidine 5'-monophosphates with phosphoric and pyrophosphoric acids as well as phosphates have given various pyro- and triphosphates belonging to the class of nucleotide coenzymes with yields ranging from 50 to 70 per cent. Represented below by way of example is synthesis of coenzyme A from the morpholide of 2',3'-cyclophosphoadenosine 5'-phosphate and pantothenoyl phosphate.
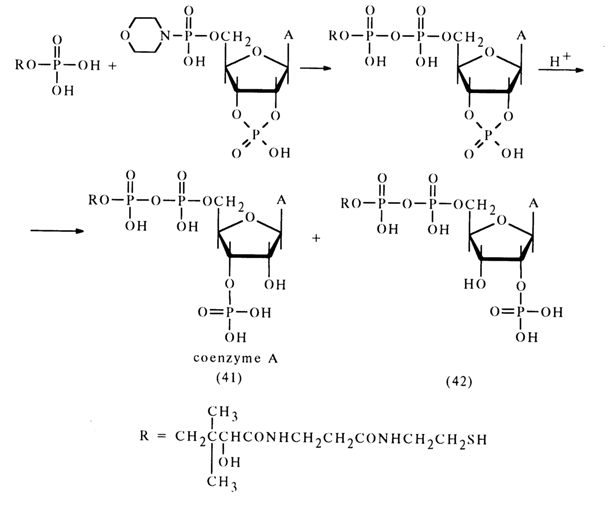
The overall yield of coenzyme (41) and isocoenzyme A (42), which are formed in equal amounts, is 65 per cent in terms of the morpholide entered into the reaction. The individually isolated coenzyme A has turned out to be identical with its natural counterpart.
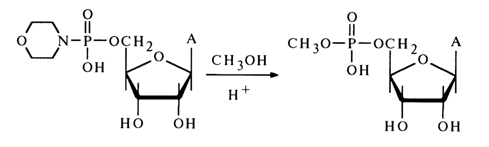
In spite of the spectacular advances in employing amidonucleotides in the synthesis of nucleotide 5'-pyrophosphates and similar compounds, attempts to use them in reactions with other nucleophilic agents have not been successful. The same is true as regards making alcohols to react with amidonucleotides to yield nucleotide esters. Only when the reaction between the morpholide of adenosine 5'-phosphate with methanol taken in a markedly (50-fold) surplus amount is conducted in pyridine in the presence of an equimolar quantity of dry HCI can a low yield of adenosine 5'-methylphosphate be achieved:
Attempts to use other alcohols in this reaction have failed. The most promising starting compounds for the synthesis of nucleotide esters are imidazolides of the latter. They have been derived from nucleotides and N,N'-carbonyl diimidazole:
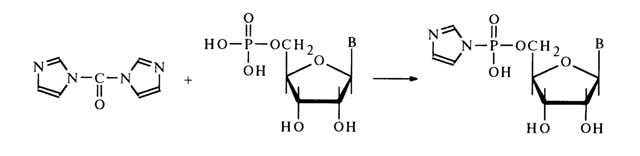
As in the case of simple phosphoimidazoles (see above), imidazolide of adenosine 5'-phosphate reacts rather easily with primary alcohols and para-nitrophenol. The reaction with tertiary alcohols proceeds with greater difficulty.