![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
4.5 Reactions of Nucleotides Involving the Phosphate Group
4.5.1 Chemical and Enzymatic Dephosphorylation
Cleavage of the phosphomonoester bond in nucleotides gives rise to nucleosides:
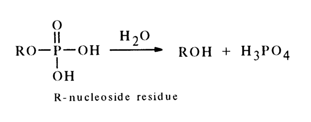
This reaction, initiated and sustained both by chemical reagents and by enzymes cleaving phosphomonoester bonds of a particular structure, is widely used in the synthesis of nucleosides and also in structural studies.
Chemical methods for hydrolysing nucleotides to nucleosides have not been studied completely enough in spite of the fact that the course of hydrolysis of the phosphomonoester bond in simple monophosphates is known in every detail. The general method of chemical hydrolysis of nucleotides to nucleosides is based on heating at 1000C in a buffer (usually ammonium formiate solution) at pH 4. As was demonstrated in experiments with simple alkyl phosphates, such conditions are optimal for cleavage of phosphomonoester bonds. The hydrolysis of nucleotides at the P-O bond seems to proceed just as in the case of simple monophosphates.
The rate of detachment of phosphate groups depends but insignificantly on their position in a nucleoside, a more important factor being the structure of the heterocyclic base. Purine nucleotides are hydrolysed faster than pyrimidine ones. For example, guanosine 5'-phosphate is hydrolysed to guanosine (pH 4, 100'C) at a rate twice that of hydrolysis of uridine 5'-phosphate to uridine.
Heating nucleotides in aqueous solutions at pH < 1 also leads to cleavage of the phosphomonoester bonds. Under such conditions, however, it is only pyrimidine nucleotides that are hydrolysed to nucleosides (cytidine being markedly deaminated in this case). When the same conditions are maintained for purine nucleotides, hydrolysis of N-glycosidic bonds proceeds at a perceptibly higher rate. Ribonucleoside 3'(2')-phosphates are readily split to nucleosides in a neutral medium in the presence of salts and hydroxides of thorium, zirconium, lead, lanthanum and some other metals. For instance, uridine 3'(2')-phosphate is almost quantitatively converted into a nucleoside when boiled for 20 min with lanthanum hydroxide:
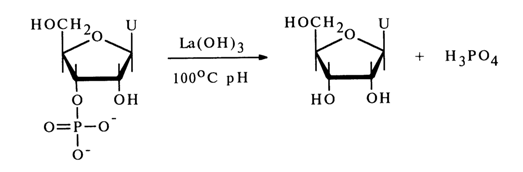
It takes two to four hours at pH 6 (1000C) for ribonucleoside 3'(2')-phosphates to be dephosphorylated in the presence of bivalent lead ions. In the presence of thorium salts at 37'C, nucleoside 3'(2)-phosphates lose inorganic phosphate both in acid (pH 3.6) and alkaline (pH 8.3) media.
Deoxyribonucleotides are not dephosphorylated under such conditions. Apparently, hydrolysis requires a hydroxyl group next to the phosphate group. The role of this group is yet to be elucidated. A possible reason for the catalysis of hydrolysis of the monoester bond by heavy metal compounds is their ability to combine with phosphate groups, which renders the phosphorus atom more electrophilic.
Enzymatic dephosphorylation of nucleotides may be mediated both by nonspecific phosphomonoesterases (phosphatases) and by phosphomonoesterases acting only on nucleotides. The nonspecific phosphomonoesterases (PME) include the alkaline phosphatase from E. coli, acid phosphatase from the prostate gland, alkaline phosphatase from wheat, and some others. The rate at which nucleotides are hydrolysed by these enzymes (to nucleosides) is usually the same as that in the case of simple monophosphates. The above phosphatases hydrolyse all mononucleotides irrespective of the phosphate group position.
Specific PME selectively hydrolyse nucleotides of a particular structure. Among the best known and widely used enzymes is that hydrolysing nucleoside 5’-phosphates. By virtue of its specific action, this enzyme has been named 5’-nucleotidase:
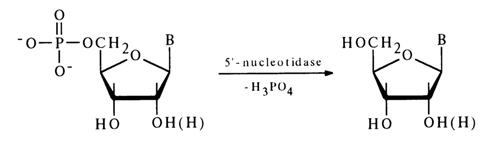
PME from snake venom is generally employed for hydrolysis of nucleoside 5’-phosphates in determining the structure of nucleotides or synthesis of nueleosides.
The phosphatases hydrolysing only nucleoside 3’-phosphates are known as 3’-nucleotidases. Such enzymes are isolated from various sources of plant origin (wheat, soybean, takadiastase, etc.). Nucleoside 2’- and 5’-phosphates are not hydrolysed by these enzymes.
4.5.2 Migration of the Phosphate Group
In the case of ribonucleoside 3'(2')-phosphates, which contain a hydroxyl next to the phosphate group, migration of the phosphate group is observed along with hydrolysis of the P-O bond in an acid medium. This process passes, just as with 1- and 2-phosphates of glycerol, through an intermediate five-membered cyclic phosphate resulting from nucleophilic substitution at the tetrahedral phosphorus atom with the participation of the neighboring hydroxyl group. Nucleoside 2',3’-cyclic phosphate is not stable in acid media and readily undergoes hydrolysis yielding a mixture of nucleoside 3’-phosphate and nucleoside 2’-phosphate:
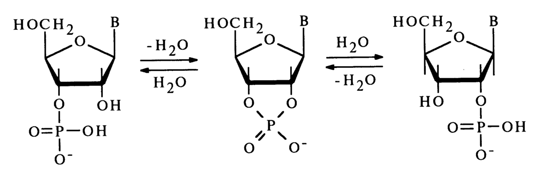
No migration takes place in alkaline media, just as in the case of glycerophosphates. To preserve pure isomers (2'- or 3’-phosphates), the ribonucleotide solution is usually alkalized with ammonia. When a mixture of isomers is separated by ion-exchange chromatography, care should be taken that the pH of the solution is above 7.
4.5.3 Alkylation of the Phosphate Group
Alkylation of nucleoside 5’-phosphates in a neutral aqueous solution with diazomethane (at 200C) yields the corresponding methyl esters of nucleotides, for example (for adenine and cytosine derivatives):
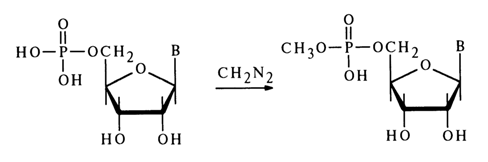
Under similar conditions, uridine and guanosine 5’-phosphates also undergo methylation at the base with N3- and N7-methyl derivatives forming along with methyl esters, for example:
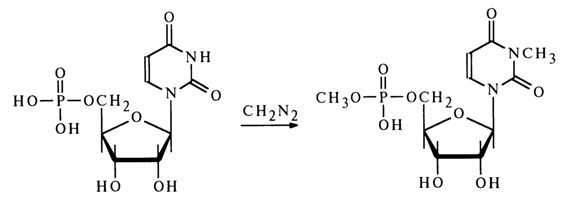
For selective alkylation of the phosphate group in nucleotides, use is made of active nucleotide derivatives.
4.5.4 Activation of the Phosphate Group in Nucleotides. Synthesis of Some Derivatives with Respect to the Phosphate Group
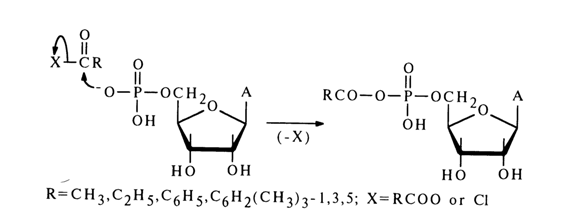
In reactions involving nucleotides, the heterocyclic base, as well as hydroxyl groups of pentose and the phosphate group may be affected. For reactions to proceed selectively with respect to the phosphate group, the latter must be activated. To this end, use is made of carbodiimides, aryl sulfochlorides, and diphenylchlorophosphate. The corresponding "active" derivatives can be obtained in the case of any nucleoside 5'-phosphates and deoxyribonucleoside 3’-phosphates. Ribonucleoside 3'(2’)-phosphates are converted, during such activation, into the corresponding ribonucleoside 2',3’-cyclic phosphates.
The resulting active nucleotide derivatives cannot be isolated because of their instability. Once formed, they are put to immediate use in the synthesis of esters, amides, anhydrides, and so forth.
Activation by Carbodiimides. Formally, carbodiimides are dehydrating agents.
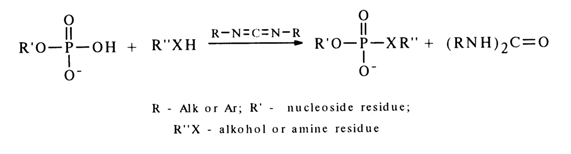
The mechanism of such reactions is yet to be completely understood, but some experimental evidence gives reason to believe that they yield a carbodiimide and nucleotide adduct (19).
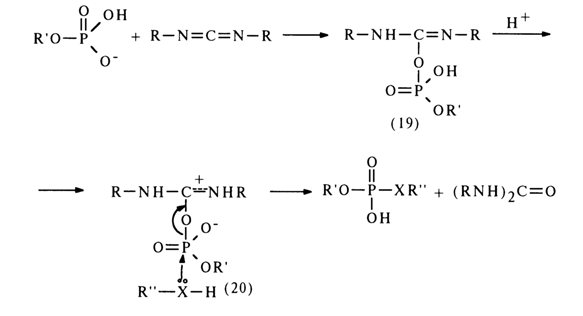
The adduct (19) subsequently receives a proton and turns into an active
derivative (20) in which the phosphorus atom becomes more electrophilic. The next step is
nucleophilic substitution at the phosphorus atom with formation of the corresponding
derivatives. NMR spectroscopic studies into the transformations undergone by nucleotides
in the presence of dicyclohexylcarbodiimide (DCC), carried out by D. G. Knorre and
coworkers, have shown that the reaction intitially yields pyrophosphates then tri- and
tetraphosphates ![]()
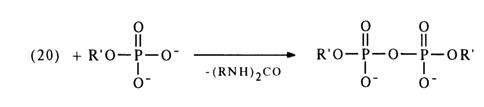
Then, the resulting pyrophosphate is activated by DCC to form a compound similar to (20), which can be converted into tri- as well as tetraphosphates.
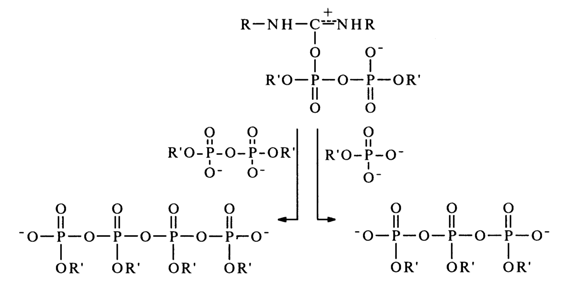
When other nucleophiles are present in the reaction mixture, the above triand tetraphosphates act as phosphorylating agents. The proposed mechanism is supported by additional experimental evidence. For instance, addition to the reaction mixture (absolute pyridine) of trialkylamines, which are strong bases and, therefore, prevent formation of protonated adducts of the (20) type, usually slows down and sometimes even inhibits the reaction. Enhancement of the nucleophilic properties of reagent R"XH speeds up the reaction perceptibly; monoalkyl phosphates (in the form of dianions), for example, enter into the reaction under consideration much more readily than dialkyl phosphates (monoanions).
Dicyclohexyl carbodiimide is often used for activation of the phosphate group. The nucleotide activated in this manner reacts smoothly with primary and secondary amines as well as various acids, giving the corresponding derivatives with respect to the phosphate group. The following scheme illustrates some examples of synthesis of nucleoside 5’-phosphate derivatives in the presence of DCC (similar transformations may also involve deoxyribonucleoside 3’-phosphates).
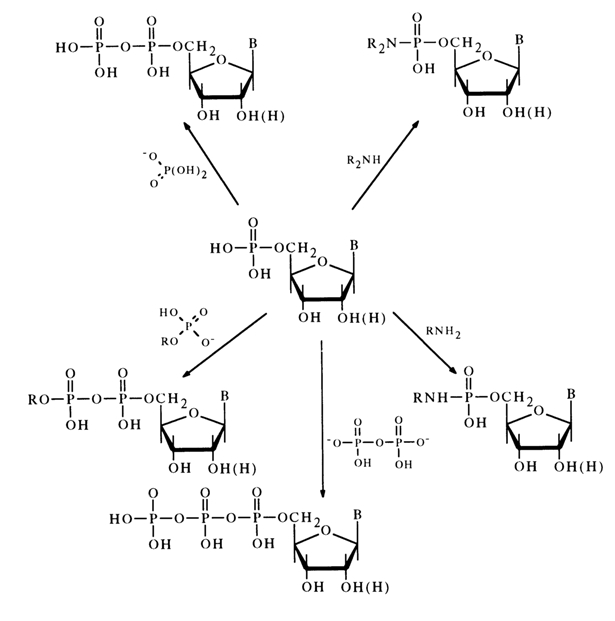
Nucleotides activated by carbodiimide can react with alcohols. In this case, in order to avoid side self-condensation reactions. the hydroxy groups of pentose and the amino groups of the heterocyclic base must be protected in advance; after condensation, the protective groups are removed with the aid of ammonia.
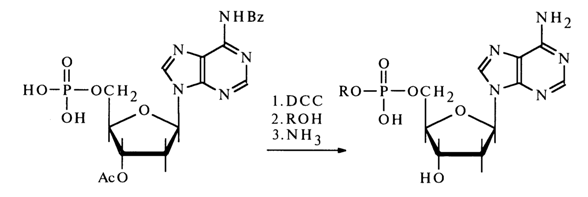
Activation by Aryl Sulfochlorides. Another method for activating nucleotides resides in their treatment with chlorides of arylsulfonic acids in absolute pyridine where nucleotides (in the form of salts with trialkylamines,) are readily soluble. The first step of the process seems to be formation of a mixed anhydride:
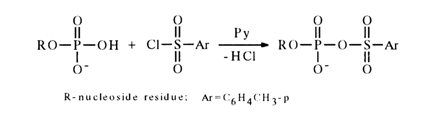
The formation of such anhydrides, however, defies detection. Knorre and coworkers have been successful in demonstrating, with the aid, of pulsed NMR spectroscopy on 31P
nuclei, that the early stage of the process is mark,ed by accumulation of pyrophosphate in the reaction mixture, along with linear trimetaphosphate: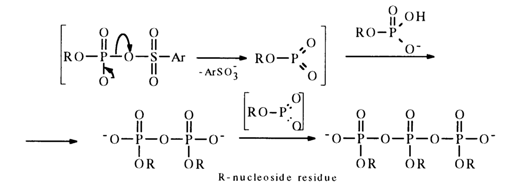
During the subsequent interaction with aryl sulfochloride the pyro- and pol.yphosphate become converted into the, corresponding monomeric nucleoside metaphosphates stabilized with pyridine (their NMR-31P spectra feature a singlet with 6 near 5 ppm), for example:
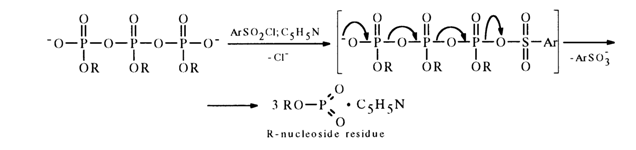
The use of an aryl sulfochloride fixed on a polymer substrate has made it possible to obtain solutions containing only a monomeric metaphosphate (the polymeric sulfonate is easily removed by simple filtration).
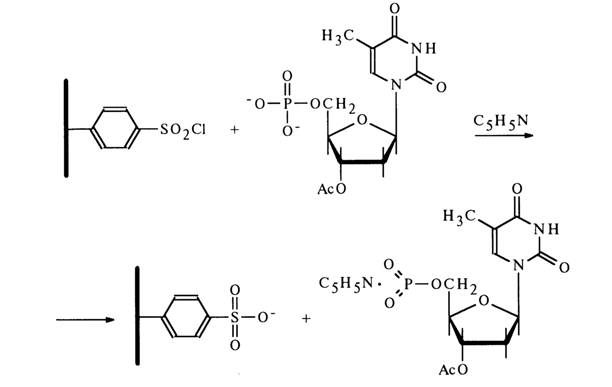
All of the reactions mentioned above are conducted under conditions of complete absence of moisture whose contact with the monomeric nucleoside metaphosphate would immediately transform it into a nucleotide:
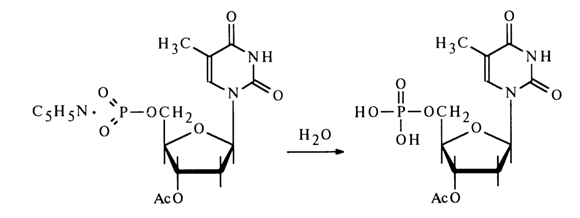
The nucleoside metaphosphates derived in this fashion can be used to obtain nucleotide derivatives with respect to the phosphate group. The following scheme represents some reactions involving 3'-acetyldeoxythymidine 5'-metaphosphate.
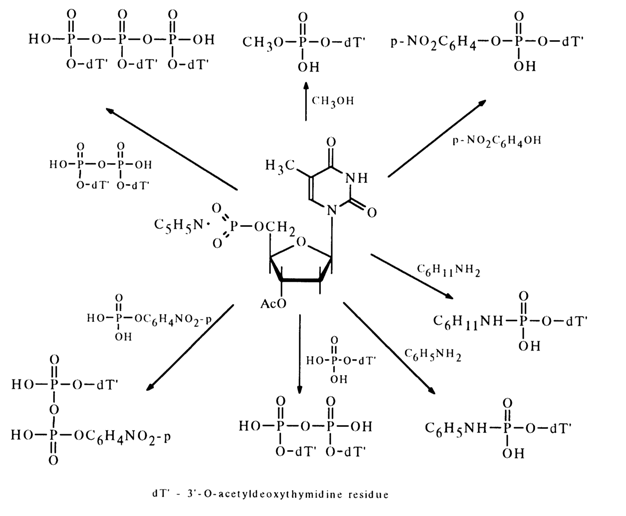
The interaction between metaphosphates and nucleosides containing a deprotected hydroxyl group in the carbohydrate moiety results in internucleotide linkage. This reaction is widely used for synthesizing oligonucleotides.
To activate nucleotides use is made of both simple aryl sulfochlorides, such as para-toluene sulfochloride, and sterically hindered ones, such as mesitylene sulfochloride (21) and 2,4,6-triisopropylphenyl sulfochloride (22):
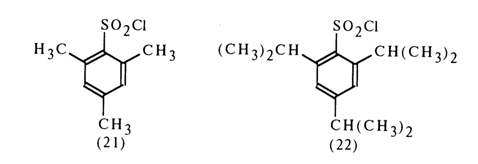
Sulfochloride (22) gives particularly good results. The reason seems to be that the leaving group forming during nucleotide activation is essentially an anion of an arylsulfonic acid which is stronger than, say, para-toluenesulfonic acid (the delocalization of the positive charge at the sulfur atom occurs virtually without participation of the aromatic ring so that the sulfo group is displaced from the plane of conjugation with the latter).
Activation by Diphenyl Chlorophosphate. The phosphate group in nucleotides is also activated with the aid of diphenyl chlorophosphate. The reaction is conducted in absolute dioxane where the forming pyrophosphate is rather stable.
Since diphenylphosphoric acid is stronger than the monophosphate (nucleotide) participating in the formation of an anhydride by the second hydroxyl of the phosphate group (pK2 ~ 6.0), in reactions with nucleophilic agents such anhydrides serve as a donor of the nucleotide (the leaving group usually is the diphenylphosphoric acid anion). Reactions of this kind proceed with particular ease in pyridine. Given below are some examples illustrating the use of a respective mixed anhydride of 5’-adenylic acid for synthesis of its esters with phenols, amides, mixed anhydrides with carboxylic acids, as well as derivatives with phosphoric acid, phosphates and similar compounds.
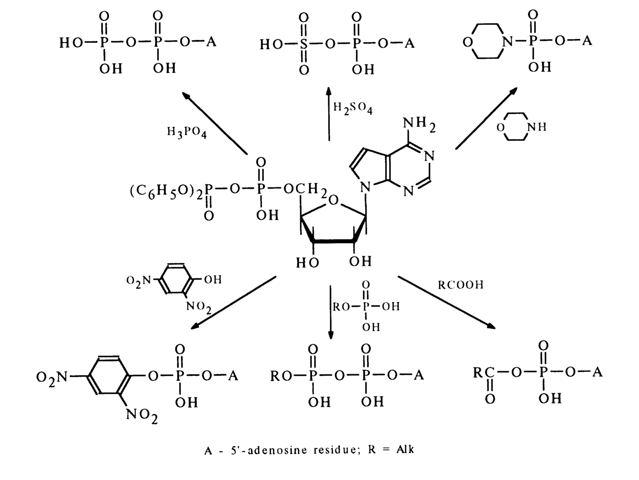
When such mixed anhydrides are employed, the hydroxyl groups of pentose are not protected because the side process of their phosphorylation both by the nucleotide and diphenyl phosphate is practically at a standstill.
Synthesis of Nucleoside Cyclic Phosphates. The above procedures for activating the phosphate group are extensively used also for intramolecular phosphorylation, for instance, synthesis of ribonucleoside 2',3’-cyclic phosphates.
When DCC is added to a solution of ribonucleoside 3'(2)-phosphate in pyridine in the presence of trialkylamine, ribonucleoside 2',3’-cyclic phosphate is produced practically with a quantitative yield:
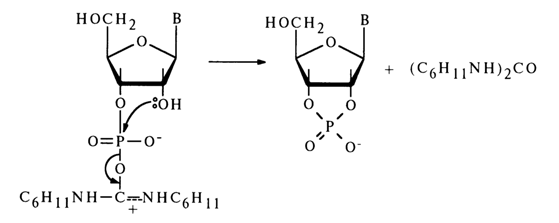
The preference of intramolecular phosphorylation leading to formation of a cyclic phosphate has been proved experimentally. It was found, for example, that when the above reaction is conducted with a large surplus of methyl alcohol (such conditions are created to synthesize methyl esters of nucleotides), no methyl ester of nucleoside 3'(2')-phosphate is formed and the only product is nucleoside 2',3’-cyclic phosphate.
Methyl esters of nucleotides, just as all diphosphates, are stable under the conditions described above and do not react even with carbodiimide.
The phosphate group can also be activated, during cyclic phosphate synthesis, by the already mentioned diphenyl chlorophosphate as well as anhydrides and chlorides of carboxylic acids, such as trifluoroacetic anhydride, chlorocarbonate, and so on. For example, addition of chlorocarbonate to an aqueous solution of uridine 2'(3')-phosphate gives uridine 2',3’-cyclic phosphate with a quantitative yield:
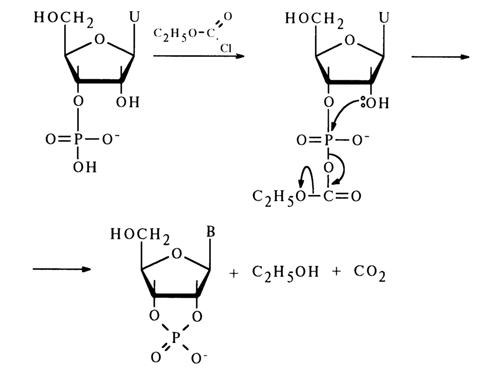
The ease of conversion of ribonucleoside 2'(3')-phosphate into a five-membered cyclic phosphate is due to the fact that the hydroxyl group in its molecule is positioned conveniently for attacking the activated phosphorus atom. During nueleophilic substitution, the hydroxyl and electron-acceptor groups can find themselves in an axial position, which will lead to formation of a new P-O bond with simultaneous departure of the electron-acceptor group:
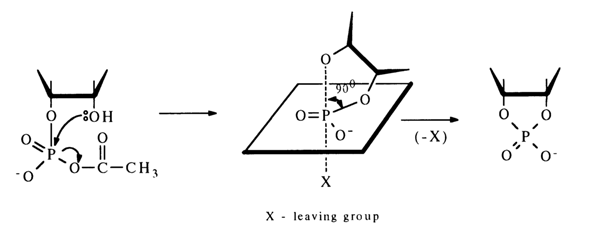
When the position of the hydroxyl group is not favorable for the intramolecular reaction, for instance, in the case of activation of nucleoside 5’-phosphates, the main process is formation of a symmetrical pyrophosphate [path (a)]:
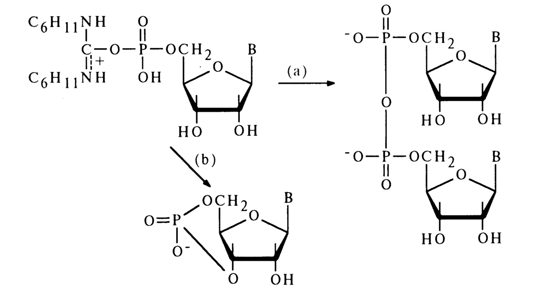
If, however, the reaction is conducted in highly diluted solutions, ribonucleoside 3',5'-cyclic phosphate is produced with a high yield [e. g., path (b)l. Thus, even a poorly positioned hydroxyl group (in transposition) can attack the phosphorus atom to form a six-membered ring.
4.5.5 Acylation of the Phosphate Group
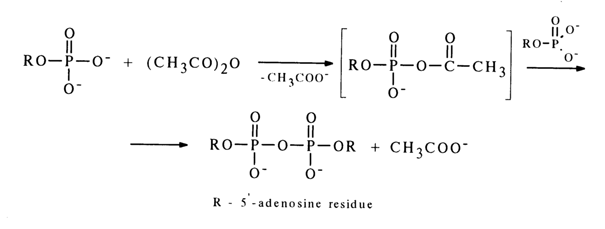
Mixed anhydrides of nucleoside 5'-phosphates with carboxylic and amino acids play a major role in many biosynthetic processes. Accordingly, methods for their synthesis have been developed. As can be seen from the scheme in section 4.5.4, by using anhydrides of nucleoside 5’-phosphates and diphenylphosphoric acid one can obtain mixed anhydrides of the corresponding nucleotides and carboxylic acids, the latter being used directly as the starting compounds. A simple and effective way to synthesize mixed anhydrides of nucleoside 5'-phosphates and carboxylic acids is based on a reaction between a nucleotide and carboxylic anhydride or chloride (for behavior of ribonucleoside 3’-phosphates in this reaction, see 4.5.4), for example:
If more than twice the excess amount of adenosine 5’-phosphate is made to react with acetic anhydride, the reaction does not terminate at the step of mixed anhydride, or acetyl adenylate, formation but yields diadenosine 5'-pyrophosphate. The latter results from nucleophilic substitution of the acetic acid anion (pKa ~ 4.8) in the intermediate acetyl adenylate by that of a weaker acid, namely, adenosine 5’-phosphate (pKa ~ 6).
Two methods are used primarily for synthesis of mixed anhydrides of nucleotides and amino acids. The first method, similar to the one described above, resides in making a nucleotide to react with a mixed anhydride of a carbobenzoxy derivative of (Cbz)-amino acid and ethyl carbonate:
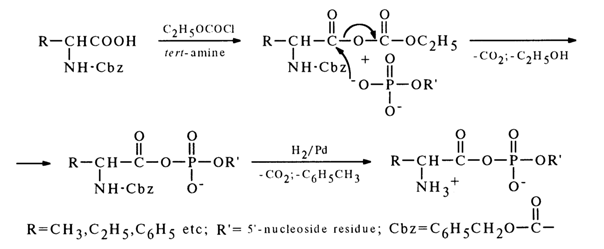
After the carbobenzoxy group is removed by hydrogenation over a palladium catalyst, the corresponding aminoacyl nucleotide is formed.
To produce mixed anhydrides of amino acids and adenosine 5’-phosphate, an alternative procedure may be used - the carboxyl group is activated in carbobenzoxy-amino acid with the aid of DCC. The resulting adduct is attacked by a nucleotide anion to give the desired mixed anhydride:
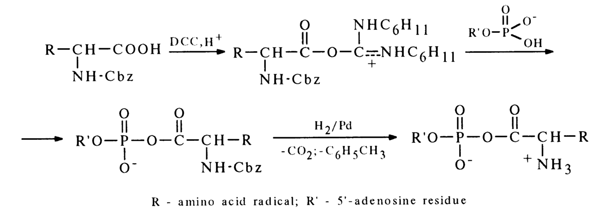
This is how a number of peptide derivatives of nucleoside 5’-phosphates were also synthesized. Moreover, DCC was also instrumental in producing anhydrides of fatty acids and adenosine 5’-phosphate.