![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
4.4 Some Properties of the Phosphate Group
(General Concepts)For a better understanding of those properties of nucleotides which stem from the presence of a phosphate group in their molecules, we must first of all review the currently adopted concepts regarding the structure of the phosphate group and also discuss, using such simple analogues of nucleotides as alkyl phosphates and similar compounds as examples, the mechanism of nucleophilic substitution at the phosphorus atom as a reaction most typical of phosphates. This will not only throw more light on certain regularities in the properties of nucleotides, but also make it possible to know more about the special properties imparted to the phosphate group by the nucleoside. In this section, the structure and properties of the phosphate group are, wherever necessary, compared with those of its formal analogue - the carboxyl group, which allows us to emphasize the peculiar behavior of organic phosphoric acid derivatives.
Moreover, alkyl phosphates are used as examples to illustrate the transformations of crucial importance in the chemistry of nucleotides and nucleic acids, namely, hydrolysis and reactions involving the neighboring groups.
4.4.1 Structure of the Phosphate Group and Mechanism of Nucleophilic Substitution at the Phosphorus Atom
As is known, the electron configuration of phosphorus is
1s2 2s2 2p6 3s2 3p3. Its valence is determined by the structure of the outermost shell which, characteristically, includes not only 3s and 3p but also 3d orbitals (Fig. 4-2).
Phosphoric acid and its esters belong to compounds with a four-coordinated phosphorus atom. Such molecules are tetrahedral. The four s-bonds with sp3
hybridization of the electron orbitals are directed toward the apices of the tetrahedron: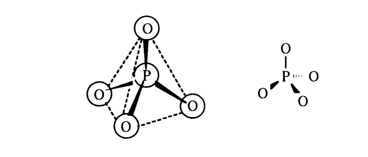
Thus, the phosphate group structure differs widely from that of the carboxyl group in carboxylic acids. It is well known that the latter group has a twodimensional structure: the formation of the three
s-bonds in it involves the sp2 -hybrid orbitals of the carbon atom (they lie in the same plane and form 1200 angles), while the fourth, p-bond results from overlapping of the p orbitals of the carbon and oxygen atoms at a right angle to this plane. The p-bond is polarized because of the pronounced electronegativity of the oxygen atom, as compared to the carbon, the degree of polarization being dependent on the inductive effect and mesomerism of the substituents at the carbonyl carbon.At present, there is no consensus regarding the nature of bonding in
compounds with a tetrahedral phosphorus atom. None the less, all assumptions to this
effect are based on the general concepts concerning the structure and properties of p
and d orbitals. It has been speculated that the p-bonding
in the phosphoryl (P=O) group may involve vacant 3d orbital ![]() . Since there are five such orbitals (see
Fig. 4-2) and each of them can, in principle, receive the fifth electron, the phosphorus
atom can interact with that of oxygen in several ways to form a p-bond in the phosphoryl group. As a result of partial
overlapping of the 3d and p orbitals of all four oxygen atoms in the
phosphate group, double bonding is not localized in the phosphoryl group but is
distributed, albeit unevenly, among all oxygen atoms of the phosphate group. For example,
knowledge of the lengths of the P-O bonds (X-ray structural data) in dibenzyl phosphate,
(C6H5CH2O)2P(O)OH, has led to a conclusion that each of the single P-O bonds accounts for
10% of double bonding and the P=O bond in the phosphoryl group accounts for 70%. Besides,
the difference between double bonds in the C = O and P = O groups lies in the fact that
the latter bond is polarized to a much greater extent (the phosphorus atom readily loses
electrons from the d orbitals and probably for this reason is less electronegative than
carbon). Such structural features of the phosphate group manifest themselves in the
greater nucleophilicity of the phosphoryl oxygen and electrophilicity of the phosphorus
atom. The transfer of electronic effects of the substituents, especially the conjugation
effect with participation of the phosphoryl group, in phosphate esters and their
derivatives is much less pronounced than in carboxylic acid derivatives because the
substituents do not lie in the same plane.
. Since there are five such orbitals (see
Fig. 4-2) and each of them can, in principle, receive the fifth electron, the phosphorus
atom can interact with that of oxygen in several ways to form a p-bond in the phosphoryl group. As a result of partial
overlapping of the 3d and p orbitals of all four oxygen atoms in the
phosphate group, double bonding is not localized in the phosphoryl group but is
distributed, albeit unevenly, among all oxygen atoms of the phosphate group. For example,
knowledge of the lengths of the P-O bonds (X-ray structural data) in dibenzyl phosphate,
(C6H5CH2O)2P(O)OH, has led to a conclusion that each of the single P-O bonds accounts for
10% of double bonding and the P=O bond in the phosphoryl group accounts for 70%. Besides,
the difference between double bonds in the C = O and P = O groups lies in the fact that
the latter bond is polarized to a much greater extent (the phosphorus atom readily loses
electrons from the d orbitals and probably for this reason is less electronegative than
carbon). Such structural features of the phosphate group manifest themselves in the
greater nucleophilicity of the phosphoryl oxygen and electrophilicity of the phosphorus
atom. The transfer of electronic effects of the substituents, especially the conjugation
effect with participation of the phosphoryl group, in phosphate esters and their
derivatives is much less pronounced than in carboxylic acid derivatives because the
substituents do not lie in the same plane.
At the same time, the phosphate and carboxyl groups share a common feature; namely, their central atom carries a partial positive charge and is therefore capable of being attacked by nucleophilic agents. When the phosphorus atom undergoes such an attack, nucleophilic substitution of one of the ligands (as the substituents at the phosphorus atom are usually called) can take place in phosphoric acid derivatives.
This process may be based on one of the two mechanisms: associative (A) and dissociative (B). In the former case, the substrate is coordinated with the nucleophile at the step determining the reaction rate. The resulting association is rather stable and may be regarded as an intermediate compound. This process is usually represented by the symbol SN2. The dissociative mechanism is characterized by the fact that the substitution rate determining step involves
departure of one of the substituents and formation of an active phosphorus compound with a smaller number of substituents at the central atom. This type of transformation is represented by the symbol SN1. In some cases, however, the substitution event is a synchronous biomolecular process and no intermediate compound is formed, in contrast to SN2 substitution at the carbon atom. The mechanism of such reactions is referred to as intermediate and denoted Ia( SNi ) ("a" indicates that the substitution process is still associative in essence). The first two mechanisms are discussed in what follows here in this section; mechanism Ia is treated in the section dealing with the properties of phosphoric acid amides whose transformations led to its discovery in the first place.
Mechanism SN2 of nucleophilic substitution at the tetrahedral phosphorus atom has been corroborated by numerous kinetic experiments with hydrolysis and alcoholysis of halogen phosphates. It has been speculated that, formally, the reaction proceeds in very much the same manner as nucleophilic substitution at the saturated carbon: the nucleophile Nu attacks the reaction site from "behind", with respect to the leaving group X.
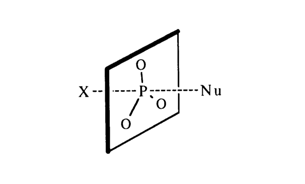
After departure of the substituent X, the configuration is turned inside out. A Walden inversion during nucleophilic substitution at the tetrahedral phosphorus atom has been demonstrated in many experiments aimed at elucidating the reactivity of optically active phosphorus-containing compounds.
It is believed that the most likely structure of the intermediate
compound is a trigonal bipyramid in which the entering and leaving (substituted) groups
(Nu and X, respectively, in Fig. 4-3A) occupy axial position ![]() .
.
Geometrically, this structure is reminiscent of the transition state arising during an S,2 reaction at the saturated carbon atom (Fig. 4-3B). In the latter case, however, the five bonds differ markedly in strength: the three bonds at the base of the bipyramid are quite strong, while the two others (C-X and C-Nu) are close to ionic bonds. At the same time, by virtue of the ability of the phosphorus atom to accept electrons at the vacant 3d orbitals (see Fig. 4-2), it can form compounds with five covalent bonds of comparable, if not identical, strength. In intermediate compounds of type A, for example (see Fig. 4-3), the three bonds at the bipyramid base (equatorial bonds), formed with the participation of sp2-hybrid orbitals, are stronger. The two other (axial) bonds involving pd-hybrid orbitals are weaker.
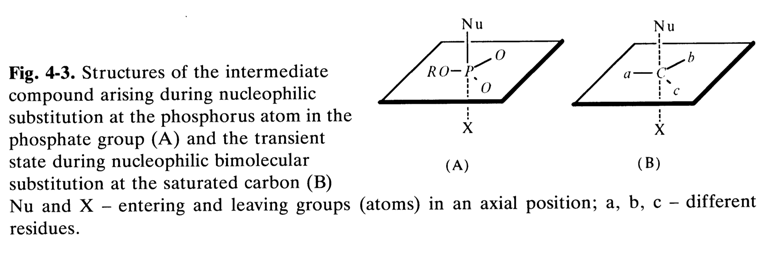
Consequently, we are dealing here with an important unique property of the phosphorus atom, namely, its ability to form additional bonds involving its vacant 3d orbitals. In this case, the octet rule is usually not observed, and the phosphorus atom in similar structures can be surrounded by a shell of ten electrons.
The mechanism of nucleophilic substitution at the phosphorus atom, described in the context of the phosphate group, differs substantially from that of nucleophilic substitution at the carbonyl carbon in carboxylic acid derivatives, where formation of the intermediate structure is ensured by rupture of the double C=O bond.
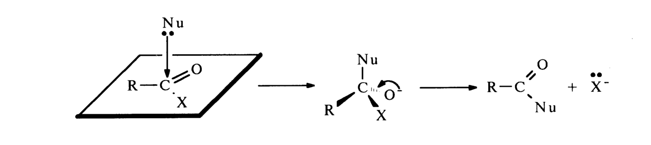
The nucleophilic attack of the carbon in the carbonyl group proceeds at a right angle to the plane accommodating the RCOX molecule. The structure emerging after the breaking of the C=0 bond is characterized by sp
3 hybridization of the electron orbitals in the carbon atom (with the four s-bonds being directed toward the apices of the tetrahedron).The rate of nucleophilic substitution at the tetrahedral phosphorus atom is determined by the following three major factors: (1) substituent species, (2) species of the leaving group, and (3) species of the nucleophilic agent.
Substituents. The ease of nucleophilic substitution at the tetrahedral phosphorus atom may be dependent both on the donor-acceptor properties of the substituents and on their three-dimensional structure.
One should begin discussion of the effect exerted by alkoxy groups in phosphates by comparing them with analogous derivatives of carboxylic acids. It is a well known fact that alkoxy groups may produce negative inductive and positive mesomeric effects on the associated atoms or groups of atoms. In cases where conjugation is possible, the positive mesomeric effect is much more pronounced than the negative inductive one.
It has already been mentioned above that carbonyl-containing compounds have two-dimensional structure. This is why the lone-pair electrons of some atoms adjacent to the carbonyl group interact with it rather strongly (are in conjugation):
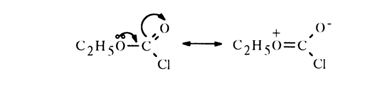
A similar conjugation in phosphates of tetrahedral configuration is not as manifest (for structural features of the phosphoryl group, see above). Nevertheless, they may also display conjugation of the oxygen and phosphorus atoms due to the vacant 3d orbitals of the latter (so-called) pp-dp conjugation). The lone-pair electrons of the oxygen in the alkoxy group may be transferred to the 3d orbitals of the phosphorus atom, which, in turn, must give rise to further hindrances for interaction between the phosphorus and the nucleophile. The presence and degree of such conjugation are confirmed by the results of hydrolysis of carboxylic and phosphoric chlorides:
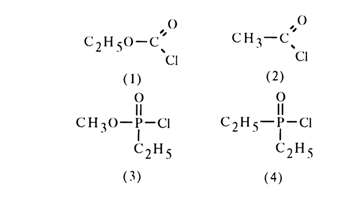
In a carbonyl compound, the substituent exhibiting p-donor properties drastically slows down the nucleophilic attack. For example, ethyl chlorocarbonate (1) is hydrolysed at a rate 104 times slower than acetylchloride (2). The effect of such a substituent is also observed in a phosphoryl compound, but to a much lesser degree. Hydrolysis of methoxyphosphoryl derivative (3), for example, is only 15 times slower than that of its alkyl analogue (4).
The fact that the p
p-dp, conjugation of substituents with the phosphorus atom affects the activity of the latter in nucleophilic substitution reactions is confirmed by well known examples of decreasing reactivity as alkyl radicals in compounds of a similar structure are substituted by alkoxy or amide groups. It has been found that reactivity during alkaline hydrolysis decreases perceptibly in the following order: (C2H5)2POCl > CH3O(C2H5)POCI > (CH3O)2POCI. The steric hindrances arising during hydrolysis of these compounds are more or less the same. Just as in the previous case, the changes in reactivity are ascribed to the fact that the lone-pair electrons of the oxygen in the alkoxy group are shifted to the 3d orbitals of the phosphorus atom. Since the 3d orbitals of the phosphorus atom are rather fully occupied as a result of such conjugation which also leads to a decrease in its surplus positive charge, incorporation of the lone-pair electrons of the nucleophilic agent into the 3d orbitals of the phosphorus atom is hindered significantly in the transition state with an associated drop in the rate of nucleophilic substitution.An even more pronounced inhibiting effect on nueleophilic substitution at the phosphorus atom is exerted by an alkylamide (or dialkylamide) group. For instance, alkaline hydrolysis of the diamide [(CH3)2N]2P(O)F proceeds at a rate slower by a factor of tens of thousands than in the case of the methylmethoxy derivative CH3O(CH3)P(O)F. This influence of the amino group in phosphamides is attributed to the fact that by virtue of its more pronounced positive mesomeric effect and less pronounced negative inductive one, as compared to the alkoxy group, it weakens the electrophilic properties of the associated phosphorus atom to a much greater degree (due to the p
p-dp conjugation).In halogen phosphates (RO)2P(O)X, the halogens (X = F and Cl) may also be in p
p-dp conjugation with the phosphorus atom. Experimental evidence attests to a lesser degree of involvement of the chlorine atom in such conjugation, in comparison with fluorine, the greater electronegativity of the latter notwithstanding (acid chlorides are much more reactive than acid fluorides). Thus, the degree of conjugation seems to be dependent not only on electronegativity, but also on the structure of the outermost shell of the atom associated with phosphorus.A similar relationship has also been observed in the case of trisubstituted phosphates and thiophosphates, (RO)2P(O)OR' and (RO)2P(O)SR', differing in the structure of one substituent. Dialkyl thiophosphates are tens of thousands of times more reactive than trialkyl phosphates.
The exact course taken by nucleophilic substitution at the tetrahedral phosphorus atom is strongly affected not only by the capacity of substituents for conjugation, but also by their size. In this case, steric hindrances are more influential than in the series of carboxylic compounds with planar structure at the carboxyl group. In esters of the (CH3O)2P(O)X type, replacement of the methyl group by a tert-butyl one leads to a rather sharp drop in the rate of alkaline hydrolysis.
Leaving Group. This factor is as important in nucleophilic substitution reactions as the electrophilicity of the phosphorus atom. The effect of the leaving group is rather simple. The greater the ease with which its linkage to the reaction site is broken, the more reactive the substance. Since the leaving group departs taking its electron pair along, all factors conducive to its greater electronegativity usually facilitate the course of the reaction. In compounds of the (RO)2P(O)X type, X is always the leaving group if it is associated with the phosphorus atom via a "weaker" bond than that linking the alkoxy group with the latter. We have already seen examples in which the X group was represented by a chlorine atom, RS and other groups. If the function of the leaving group is performed by a phosphoric acid anion, when nueleophilic substitution takes place in pyrophosphates, for example, the attack ends in liberation of a more stable anion - that is, anion of a stronger acid. For instance, asymmetrical dibenzyldiphenyl pyrophosphate reacts with alcohols to yield exclusively dibenzyl phosphates (diphenylphosphoric acid is stronger than the dibenzylphosphoric one).
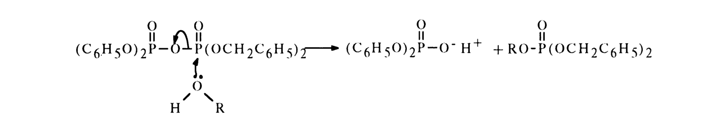
Nucleophilic Agent. In the course of nucleophilic substitution. the nucleophile displaces the group being substituted from the molecule of the substrate (in our case, phosphate). As has already been pointed out, the ease with which the nucleophilic substance is attached to the phosphorus atom acting as the reaction site depends on a number of factors. One of them is nucleophilicity or, in other words, capacity of a reagent to give up its electron pair for combining with the reaction site. Nucleophilicity is determined by the mobility of electrons in the nucleophilic particle, its polarizability, and steric factors. The nucleophilic agents reacting readily with the tetrahedral phosphorus atom can be arranged as follows [with respect to chlorophosphinates of the R2P(O)CI type]: HO- > C6H5O-
> C2H5OH > C2H5S-, CH3COO-. Much more pronounced nucleophilicity is displayed by hydroxylamine and hydroxamic acids. The increased activity of these compounds stems from the fact that the atom involved in the nucleophilic attack is found next to another having lone-pair electrons ("a-effect"). This seems to ensure greater polarizability of the nucleophilic agent. Naturally, depending on how fully the 3d orbitals of the phosphorus atom are occupied as a result of pp-dp, conjugation with the substituents. the basicity and polarizability of nucleophiles may influence the reaction rate differently. It has been demonstrated that the more pronounced the conjugation of substituents, for example, alkoxy and dialkylamide groups with 3d orbitals, the less significant the effect of basicity of the nucleophile on the reaction rate and the greater the importance of polarizability.It has already been mentioned that during substitution based on the SN2 mechanism in the transition state, the bonds of the saturated carbon atom (see Fig. 4-3B) differ widely in strength, and the transition state is not likely to exist for a more or less long time. At the same time, the corresponding state for the phosphorus atom in phosphates (see Fig. 4-3A) is characterized by a much smaller difference in bond strength and is comparatively more stable. It is these specific features that make restructuring of the transition state (type A in Fig. 4-3) possible, which may ultimately affect the end result of the substitution. Such restructuring is not accompanied by cleavage of the phosphorus-substituent bonds and boils down to changes in bond angles and lengths in a sufficiently persistent transient state, as represented by the trigonal bipyramid. This kind of restructuring is schematically shown below.
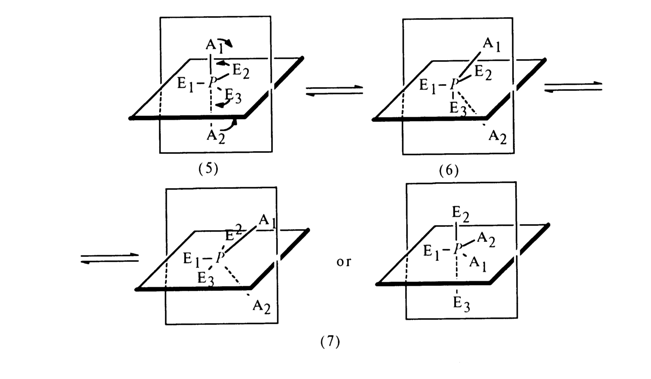
Let substituents E1, E2
and E3 in the starting trigonal bipyramid occupy equatorial positions, with substituents Al and A2 being in axial ones.At a certain point in time, the substituents A1 and A2
start moving toward each other in the plane of the page. Simultaneously, the substituents E2 and E3 move in a plane perpendicular to that of the page so that E3 moves upward from the page, while E2 moves in the opposite direction. As a result, a state (6) arises in which the substituents A1, A2 and E3 form the base of a tetragonal bipyramid with apex E1. The subsequent changes in angles, proceeding in the same directions, lead to a state (7) where the substituents E3 and E2 lie on the same straight line, whereas El, A1, and A2 are arranged in the plane of the page at an angle of 1200. In the resulting trigonal bipyramid, the axial positions are now occupied by the substituents E3 and E2 and the equatorial positions are occupied by El, A1 and A2. Such restructuring of the transient state, (5) ® (7), which, of course, is reversible, is called pseudorotation. The line connecting the central atom with the substituent El is known as axis of rotation. The pseudorotation phenomenon is observed in such compounds where the energies of the phosphorus-substituent bonds are reasonably close. In the state (6), the bond lengths are practically equalized (rehybridization of the three sp2 equatorial and two pd axial orbitals, leading to five sp3d hybrid orbitals), and the function of the axis of rotation can be performed by virtually any phosphorus-substituent bond, provided it does not differ markedly from others. Some examples of pseudorotation are considered below in section 4.4.4 dealing with cyclic phosphates.Mechanism SN1. Some reactions of nucleophilic substitution at a tetrahedral phosphorus atom are believed to involve a mechanism different from SN2 described above. The reaction apparently proceeds in two steps, in this case: first, one of the substituents (X) in the phosphate group is eliminated with intermediate formation of the corresponding metaphosphate, then, the nucleophilic agent (Y - ) is attached to it:
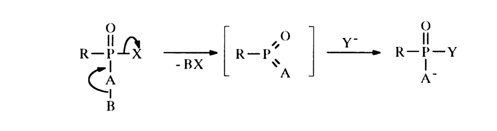
This is what is known as mechanism SN1 of phosphorus substitution. However, the existing proof of the latter cannot yet be considered convincing enough. Now follow examples of reactions believed to be based on this mechanism.
Salicyl phosphate (8) is hydrolysed much more rapidly than unsubstituted phenyl phosphate or para-carboxyphenyl phosphate (the hydrolysis rate is maximum at pH 5). It was assumed earlier that nucleophilic catalysis takes place and salicyloyl phosphate (9) undergoes hydrolysis. As was established later. the reaction takes a different course: salicyl phosphate (8) is hydrolysed more rapidly than salicyloyl phosphate (9) and is essentially an intermediate compound forming during hydrolysis of the latter:
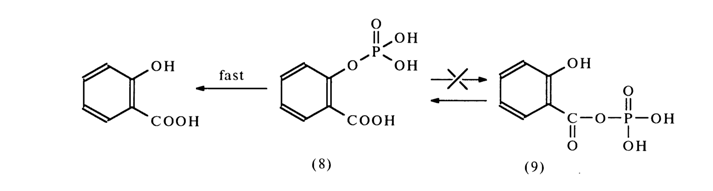
The reason for a faster reaction rate in going from (9) to (8) seems to be intramolecular acid catalysis leading to formation of metaphosphate [PO3-]:
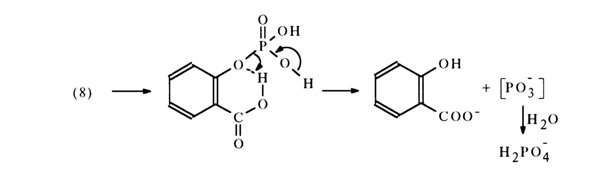
1,8-Dihydroxynaphthalene monophosphate is hydrolysed in a similar manner:
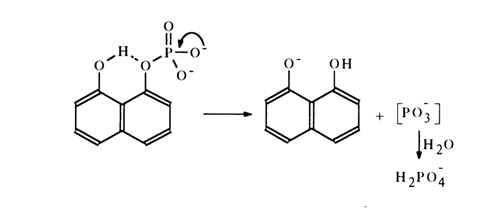
The hydrolysis of acyl phosphates and pyrophosphates also seems to involve the SN1 mechanism:
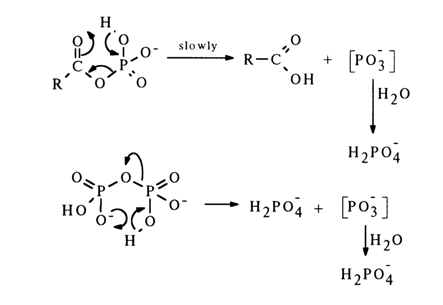
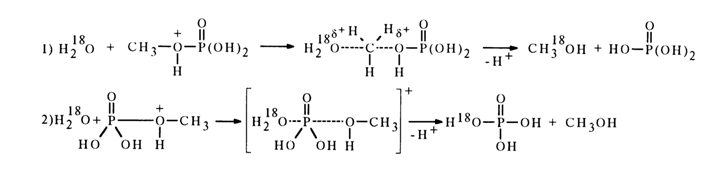
More substantial evidence in favor of the SN1 mechanism of substitution at the phosphorus atom was provided in studies of tetraethyl pyrophosphate hydrolysis. The reaction proceeds at a fast rate at pH values exceeding 9 and is catalyzed by phosphate ions. If the catalyst is a phosphate ion containing the oxygen isotope 18O
, the label is detected in the molecule of the emerging diethyl phosphate. This is indicative of intermediate formation of a metaphosphate anion in this reaction.This conclusion is borne out by formation of methyl phosphate (11) when the reaction is conducted in methanol and also trimetaphosphate (10) in a concentrated aqueous solution.
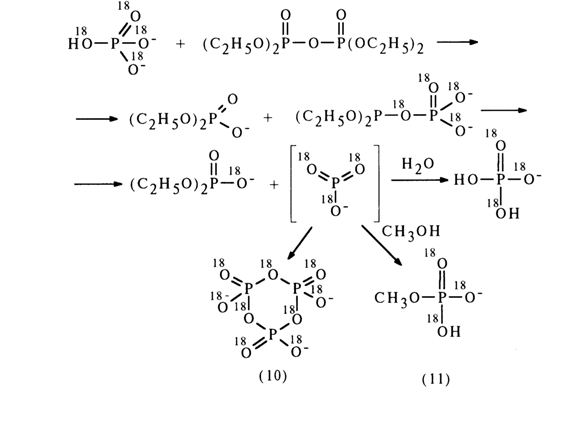
4.4.2 Catalysis of Nucleophilic Substitution at the Phosphorus Atom
Nucleophilic Catalysis (increasing the reactivity of the phosphorus atom). Many reactions with the participation of phosphate derivatives have been found to involve nucleophilic catalysis by tertiary amines. Alcoholysis of tetrabenzyl pyrophosphate, for example, is accelerated by imidazole:
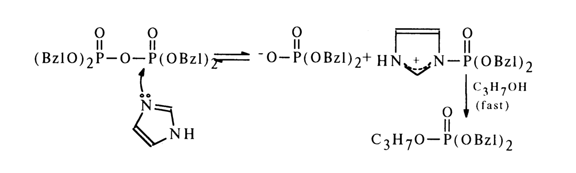
A similar effect is produced by pyridine, however, sterically hindered amines, such as 2,6-lutidine, do not catalyze such reactions.
Catalysis with a nucleophilic agent resides in that substitution of the leaving group with formation of an intermediate compound and subsequent replacement of the catalyst by the nucleophile whose entry is facilitated by it proceed at much faster rates than in a non-catalyzed reaction. This has the following explanation: tertiary amine is a stronger nucleophile than alcohol, and the forming intermediate with an ammonium group at the phosphorus atom is more reactive than pyrophosphate because the p
p-dp, conjugation between the phosphorus and substituent containing the ammonium nitrogen is completely upset and the latter exerts a strong negative induction effect. An example of unstable compounds belonging to this category is provided by pyridinium derivatives of alkyl phosphates: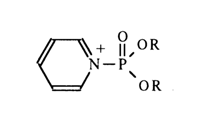
General Basic Catalysis. What sets this type of catalysis apart from the nucleophilic one is the deuterium isotope effect. It is known that reactions whose rate is determined by cleavage of the bonds formed by deuterium atoms proceed five to eight times more slowly than the corresponding reactions involving atoms of protium (instead of deuterium). Alcoholysis of pyrophosphates in the presence of bases is slowed down from ROH to ROD. This indicates that during the step preceding nucleophilic attack at the phosphorus atom the alcohol is activated by the base, the activation boiling down to "removal" of the proton:
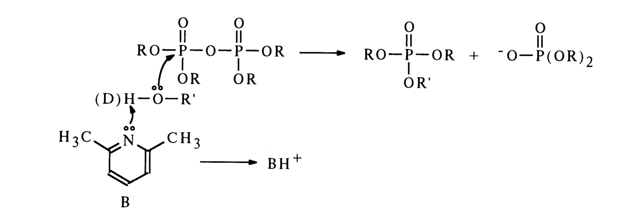
Since in this case the base serving as the catalyst does not participate in the nucleophilic attack at the phosphorus atom, its steric features do not affect the reaction rate in any significant manner. For instance, phosphorylation of alcohols with pyrophosphates (see above) is catalysed by tertiary amines, including such sterically hindered ones as, say, 2,6-lutidine.
General Acid Catalysis. A good illustration of acid catalysis of nucleophilic substitution at the phosphorus atom is provided by reactions between sarin (isopropyl methylphosphonofluoridate) with phenols. For example, the pyrocatechol monoanion reacts with sarin at a much faster rate than the phenolate ion.
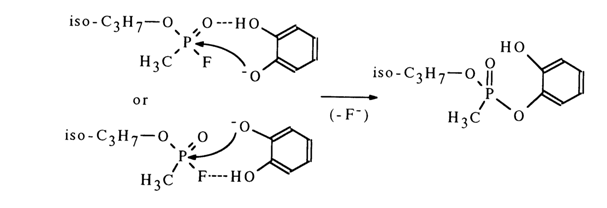
A possible reason is hydrogen bonding between the nondissociated hydroxyl group of pyrocatechol and the phosphoryl oxygen or sarin fluorine atoms, disturbing the oxygen-phosphorus conjugation with the result that the phosphorus atom is more readily attacked by the nucleophile, or phenoxy ion to be precise. Catalysis along similar lines seems to be likely for reactions in which the function of the catalytically active group is performed by the hydrogen-containing ammonium group.
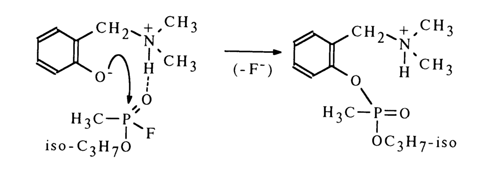
The exceptionally high rate of acid hydrolysis of a sarin analogue containing a trialkyl amino group can be ascribed to intramolecular catalysis.
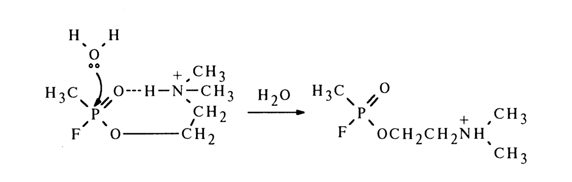
The general acid catalysis may involve nondissociated carboxyl groups. Diethyl phenylphosphonate which contains a carboxyl group in the ortho position is easily hydrolysed, when heated with water, to a free phosphate:
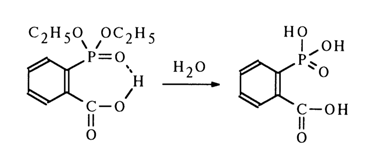
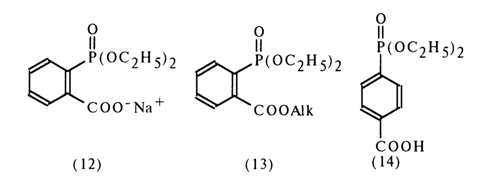
The stability toward hydrolysis under the same conditions of the sodium salt (12) and alkyl esters (13) of the same acid as well as the corresponding paraisomer (14) provides additional proof to support intramolecular acid catalysis with the participation of an ortho-carboxyl group.
Electrophilic Catalysis. Many examples may be used to illustrate catalysis of phosphate hydrolysis by metal ions. A case in point is alcoholysis of tetrabenzyl pyrophosphate with a propyl alcohol and tertiary amine mixture, the rate of which substantially increases in the presence of lithium or calcium ions.
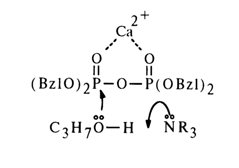
This effect also seems to be a consequence of enhanced electrophilic properties of the phosphorus atom after drawing electrons from the phosphoryl oxygen. Hence, the mechanism of catalysis in this case is close to the acid one, with the difference that the role of the proton is played by the metal ion. Similarly, diisopropyl fluorophosphate and related compounds are rapidly hydrolysed in the presence of complexes formed by bivalent copper and
a,a'-dipyridyl: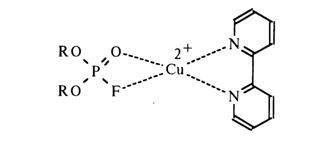
4.4.3 Hydrolysis of Alkyl Phosphates
Since nucleotides and polynucleotides remained for a long time relatively little known compounds, most conclusions made as regards the mechanisms of their transformations associated with the phosphate group more often than not were based on analogies with the corresponding reactions involving alkyl phosphates. In the early fifties, for example, evidence was provided, by the mechanisms of hydrolysis of mono- and dialkyl phosphates as well as phosphates of simple polyols, to explain the difference in hydrolytic stability between RNA and DNA. The same findings made it possible to predict and explain many properties of nucleotide derivatives from the structure of the associated nucleoside and the phosphate group position in the latter. The facts accumulated so far are not yet sufficient for detailed analysis of the mechanisms of many reactions involving nucleotides and their derivatives, especially enzymatic reactions with participation of the phosphate group. There is no doubt, however, that a major contribution to investigation of these processes will be made by the thoroughly studied reactions involving such analogues of nucleotides as alkyl phosphates and phosphates of polyols. Accordingly, this section deals with reactions involving mono-, di- and trialkyl phosphates and also those of polyols. The reactions discussed here are of great importance in the chemistry of nucleotides and their derivatives. Particular attention is given to processes with participation of the neighboring groups - that is, accompanied by intramolecular catalysis. Most of the reactions considered in this section are based on the mechanism of nucleophilic substitution at the tetrahedral phosphorus atom. However, depending on the exact form in which the phosphate group takes part in the reaction (anion, neutral molecule), the latter may take a different course as well. The general concepts as regards such reactions are also discussed.
Hydrolysis of Monoalkyl Phosphates. The rate of hydrolysis of most simple monoalkyl phosphates passes through a maximum lying in the pH range of 3 to 5. A typical curve showing the alkyl phosphate hydrolysis rate versus pH of the medium is given in Figure 4-4.

At alkaline pH values, the hydrolysis rate drops sharply. At acid pH values, a minimum is observed (at pH 1), then the hydrolysis rate increases again with acidity of the medium. Since the pKa values for the first and second steps of dissociation of methyl phosphate are 1.54 and 6.31, respectively, it can be assumed that the hydrolysis rate maximum at pH 4 corresponds to the highest monoanion concentration in the solution. The decreasing hydrolysis rate in an alkaline medium suggests that the dianion is not active (apparently because the electrostatic repulsion hinders the attack of the dianion by the hydroxyl ion). A nondissociated acid is less active than its monoanion. At pH < 1, one can observe acid-catalyzed hydrolysis with formation of a conjugate acid:
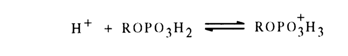
The mechanism of hydrolysis of the methyl phosphate monoanion has been at the center of protracted debates which ended in rejection of the bimolecular substitution hypothesis (SN2). The methyl phosphate monoanion hydrolysis rate is much higher than that for the dimethyl phosphate monoanion (see below). This has led to speculations that for a molecule to display some kind of "special" reactivity, in a substituted phosphate a nondissociated hydroxyl must coexist with a negatively charged oxygen atom. Two very similar S,1 mechanisms of general intramolecular acid catalysis with participation of the nondissociated hydroxyl group in the monoanion have been discussed. It has been assumed that the proton can be transferred to the leaving group either with involvement of water, through emergence of a six-membered cyclic transition state, or without such involvement, through intramolecular migration:
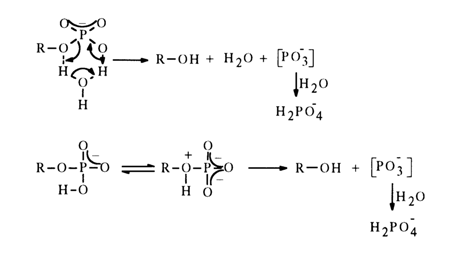
Both mechanisms include formation of a hypothetical metaphosphate. There is no direct proof of metaphosphate formation during hydrolysis of monoalkyl phosphates at pH ~ 4, yet the hypothesis is supported by some indirect evidence. The stability of monophosphate dianions, if their hydrolysis is assumed to be based on a similar SN1 mechanism, seems to stem from the fact that many alkoxy groups are eliminated but with difficulty and can "depart" only if they are protonated in advance, as is the case with monoanions. Dissociation in an alkaline medium with formation of an RO- anion occurs only in the case of esters containing strong electron-acceptor groups for example:
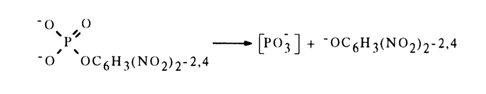
In spite of the many difficulties arising in the investigation of hydrolysis of the neutral form of monoalkyl phosphates, ROPO3H2
, it was found to be accompanied by rupture of the oxygen-carbon bond in the R radical. Thus, in this case the monophosphate acts as an alkylating agent, just as alkyl sulfates. The substitution proceeds as an SN2-type reaction at the saturated carbon atom: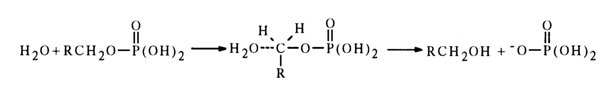
As regards tert-butyl phosphate and
a-D-glucose 1-phosphate, whose neutral forms are hydrolysed at a rate 105 times fasterThe mechanism of hydrolysis of such esters seems to be determined by the ease of formation of the intermediate carbocation.
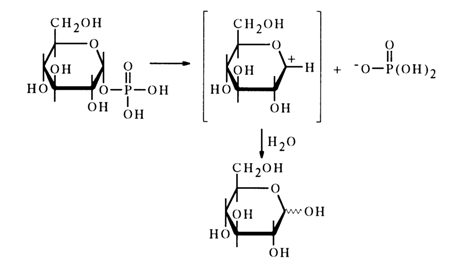
The hydrolysis of monophosphates of primary alcohols at pH 1 is based on a bimolecular mechanism. Experiments with H218 0 have shown that two parallel processes occur. The first process involves cleavage of the C-0 bond (73 %), while in the second process it is the P-O bond that is broken (27 %):
Monophosphates formed by secondary and tertiary alcohols are hydrolysed in an acid medium by a monomolecular mechanism with cleavage of the C-O bond only, for example:
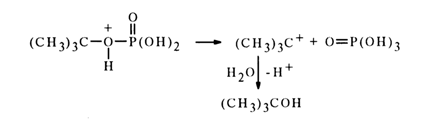
Table 4-2 is a brief summary of the effect of molecular structure of monoalkyl phosphates on their behavior during hydrolysis.
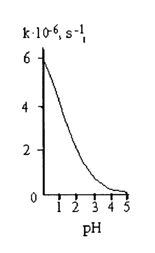 Fig.
4-5. Dimethyl phosphate hydrolysis rate versus pH of the medium at 1000C
Fig.
4-5. Dimethyl phosphate hydrolysis rate versus pH of the medium at 1000C
Table 4-2. Effect of Molecular Structure of Monoalkyl Phosphates on Their Behavior During Hydrolysis.
Hydrolysis of Dialkyl Phosphates. Dialkyl phosphates are among the
least reactive esters ![]() . The curve representing their hydrolysis
rate constant as a function of pH has no maximum (Fig. 4-5), which is indicative of the
extremely slow hydrolysis of the dialkyl phosphate monoanion.
. The curve representing their hydrolysis
rate constant as a function of pH has no maximum (Fig. 4-5), which is indicative of the
extremely slow hydrolysis of the dialkyl phosphate monoanion.
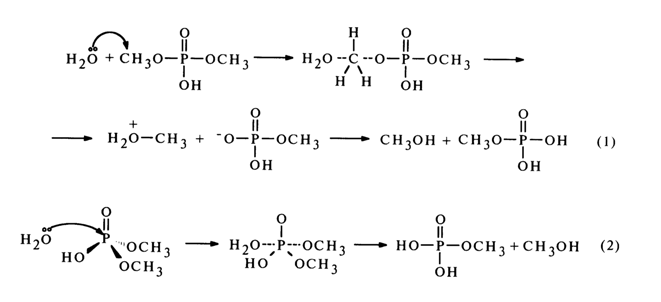
Hydrolysis of the diester in its neutral form proceeds predominantly (70-80%) with cleavage of the C-O bond [equation (1)], while the P-O is broken only in 20 to 30 per cent of the cases [equation (2)].
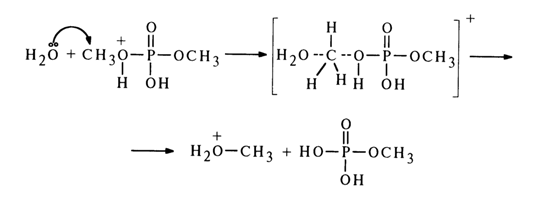
As the medium becomes more acidic, the dialkyl phosphate hydrolysis rate increases with acid concentration. The protonated form of the diester is hydrolysed, primarily, at the C-O bond (up to 90%), just as the neutral form, the P-O bond being affected insignificantly (10 %):
Thus, the widest difference in the hydrolysis rate of di- and monophosphates is observed in the case of the monoanionic form. The differences in the rates of hydrolysis of other ionic forms are not as pronounced.
Hydrolysis of Trialkyl Phosphates. Triphosphates undergo hydrolysis in both acid and alkaline media. At alkaline pH values, the mechanism of the reaction is bimolecular and includes a nucleophilic attack of the tetrahedral phosphorus atom by a hydroxyl ion (experiments with H218O have shown that the P-O bond is usually broken in this case):
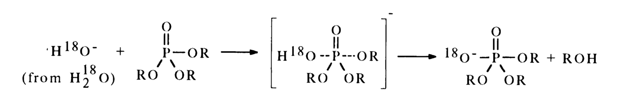
The geometric structure of the transition state is a trigonal bipyramid in which the incoming and leaving groups occupy axial positions. This is corroborated by the fact that alkaline hydrolysis of optically active triphosphates is usually accompanied by inversion. Formation of the transition state involves polarization of bonds - the nueleophile (anion) donates electrons to the phosphorus atom. When the substituent departs in anionic form, the phosphorus loses these electrons and passes into the initial state. Due to the high sensitivity of the conjugation toward charge reversal, it is more pronounced in the transition, as opposed to the ground, state. In the presence of an RO group with a negative inductive effect in the triester molecule, two effects opposite in sign are observed, namely: inductive effect, leading to displacement of a electrons from the phosphorus atom along the P-O bond (in the P-O-R group), and an increase in the p
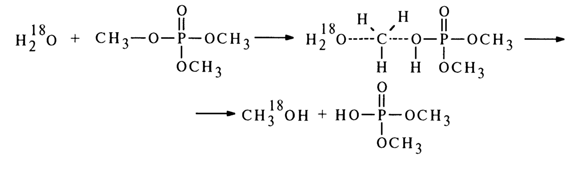
p-dp, conjugation, with displacement of the lone-pair electrons of the oxygen in the RO group to the 3d orbitals of the phosphorus atom. As the electron-donor property of the radicals (R) in the triphosphate becomes more pronounced, the positive mesomerism of the RO groups increases, which leads to shorter P-O bonds in the P-O-R groups and a much slower rate of alkaline hydrolysis.In neutral and acid solutions, the hydrolysis rate for triphosphates is weakly dependent on the proton concentration, which suggests that the unprotonated form of the phosphate is hydrolysed at a faster rate. Experiments with H2180 have shown that it is the C-O bond that is broken predominantly in an acid medium:
4.4.4 Cyclic Phosphates
Hydrolysis of Cyclic Phosphates.
Unlike simple dialkyl phosphates, fivemembered cyclic diphosphates are extremely reactive. Ethylene phosphate, for example, is hydrolysed with extreme case in both acid and alkaline media. Under such conditions, the hydrolysis rate for this compound is about 107 or 108 times faster than that for dimethyl phosphate (see above), and only the P-O bond is broken.The high reactivity of cyclic phosphates, as compared to non-cyclic ones, can be
explained by the ring being strained. The straining seems to occur only in five-membered
cyclic phosphates, in view of the fact that the reactivity of compounds with a larger
(six- and seven-membered) ring equals that of dimethyl phosphate or is even lower. The
exact nature of the conjugation in fivemembered cyclic phosphates is yet to be elucidated,
however, the greater ease of their hydrolysis, in comparison with acyclic derivatives,
suggests that formation of the transition state is accompanied by elimination of the
strained state ![]() . This hypothesis is supported by the fact
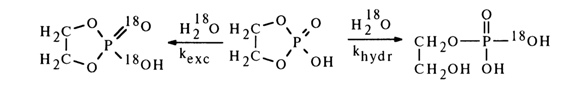
that treatment of ethylene phosphate with 18O-labelled water in an acid medium
along with incorporation of the isotope into the phosphate group as a result of hydrolysis
also leads to oxygen exchange without breaking of the ring, the latter reaction proceeding
at the same rate as hydrolysis (khydr/kexch =5).
. This hypothesis is supported by the fact
that treatment of ethylene phosphate with 18O-labelled water in an acid medium
along with incorporation of the isotope into the phosphate group as a result of hydrolysis
also leads to oxygen exchange without breaking of the ring, the latter reaction proceeding
at the same rate as hydrolysis (khydr/kexch =5).
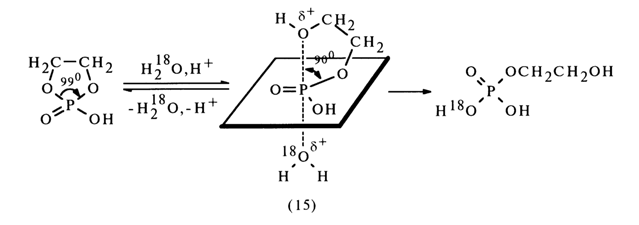
Consequently, emergence of a transition state has been postulated for both reactions, having the structure of a trigonal bipyramid (15) (see below) in which the P-O bonds of the five-membered ring occupy an axial position and an equatorial one. In this nonplanar ring, the O-P-O angle is 900, as opposed to 990 in the starting cyclic phosphate.
Calculations of the angular straining have shown that the gain in energy. resulting from elimination of the state of strain during emergence of the transition state (15), ranges from 12 to 25 kJ/mole (3-6 kcal/mole). In accordance with the rules of nucleophilic substitution at the tetrahedral phosphorus atom, the incoming and leaving groups occupy axial positions. Consequently, hydrolysis of ethylene cyclic phosphate must be accompanied by cleavage of the P-O bond in the axial position as well as by formation of hydroxyethylene phosphate containing an oxygen isotope in the phosphate group:
On the other hand, oxygen exchange in ethylene phosphate with the fivemembered ring remaining intact must also occur in keeping with the general rule - that is, departure of the group being substituted (in this case, hydroxyl) from the axial position. Hence, in the transition state of type (15), resulting from nucleophilic substitution in the ethylene phosphate molecule, the axial positions may be occupied not only by -O-CH2- groups but also hydroxyl ones with labelled and unlabelled oxygen atoms. This may also occur as a result of restructuring of the transition state, known as pseudorotation. In the case under consideration, the corresponding transformation can be written as follows:
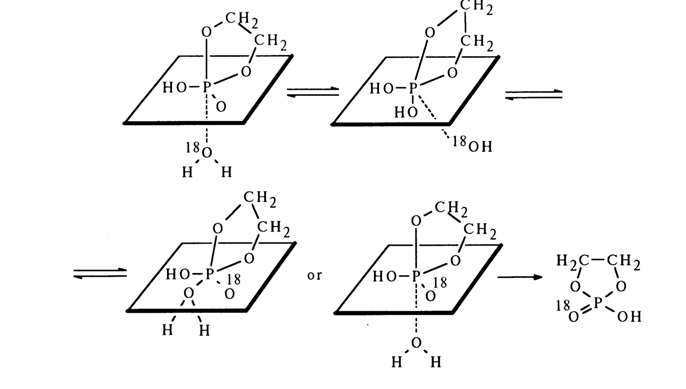
It can be seen that the oxygen exchange may occur without opening of the ring. The probability of another possible restructuring of the starting trigonal bipyramid, during which the O-P-O group finds itself in the equatorial position, is low because in such an event the O-P-O angle would be equal to 1200 and that would render the system much more strained.
A similar mechanism also seems to be involved in hydrolysis of methylethylene cyclic phosphate which has been found to undergo not only rupture of the cycle and oxygen exchange but also rapid hydrolysis of the exocyclic group with the ring remaining intact.
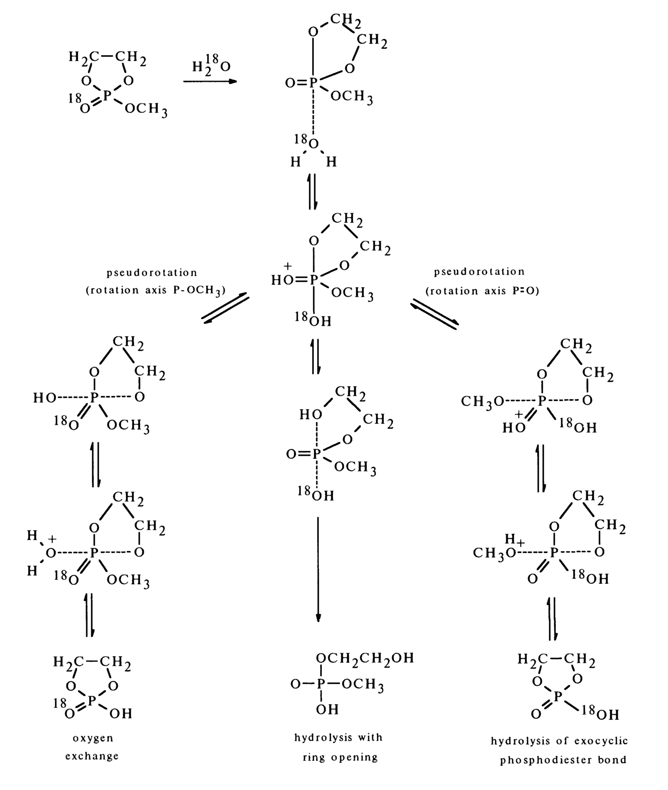
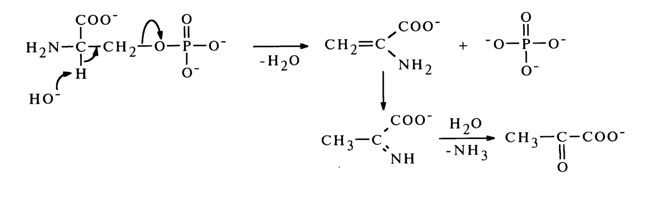
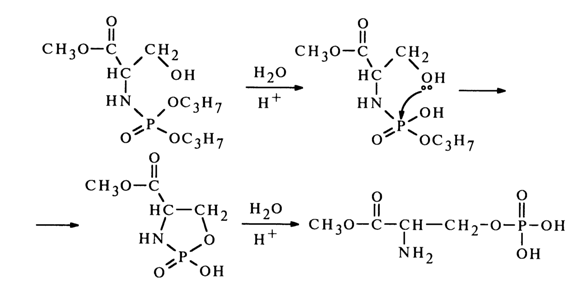
Reactions Accompanied by Formation of Cyclic Structures Including a Phosphate Group. It is known that in an acid medium the phosphate group in N-phosphorylated hydroxyamino acids undergoes N ® 0 migration. For example:
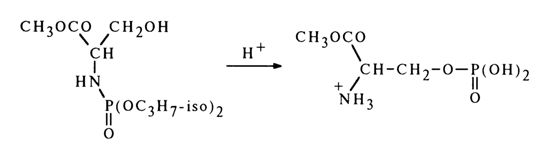
In this case, one might expect emergence of a transition state incorporating a five-membered ring:
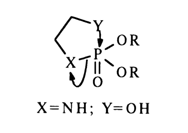
However, the three constituent atoms in the resulting five-membered ring must lie on the same straight line (the leaving group X and incoming group Y must be in an axial position or, in other words, the X-P-Y angle must be 1800), which is impossible. Since such migration causes detachment of both isopropyl groups, it is more likely to expect a sequence of steps including hydrolysis and cyclization with departure of the isopropoxy groups and opening of the ring:
A similar migration of the phosphate group was observed during alkaline treatment of glycero-1-methyl phosphate (16):
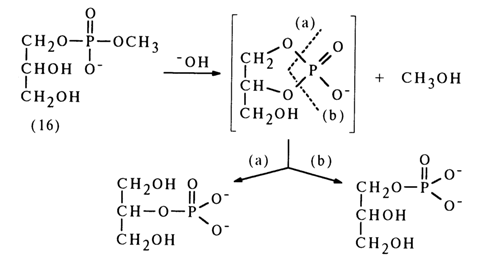
The formation of equal amounts of glycerol I- and 2-phosphates is due to emergence of a five-membered cyclic phosphate as a result of the nucleophilic attack on the phosphorus atom by the adjacent hydroxyl. As has already been mentioned, such compounds are highly unstable and undergo hydrolysis at virtually the same rate along both possible paths (a) and (b).
Methyl(b-hydroxyethyl) phosphate is fully converted into hydroxyethyl phosphate in a similar manner:
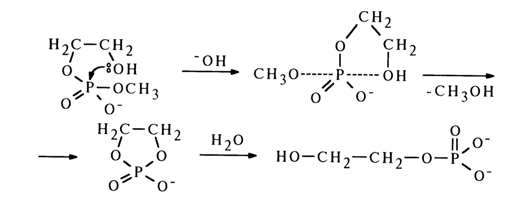
Under the same conditions, methyl(
b-methoxyethyl) phosphate, which cannot be transformed to a cyclic structure, is not hydrolysed. The above findings suggest that hydrolysis of diphosphates rather stable in an alkaline medium (see above) is substantially facilitated in cases where cyclic phosphates can form as intermediate products of hydrolysis.The fact that alkaline hydrolysis of glycero-1-methyl phosphate fails to produce glycero-2-methyl phosphate, the main process being elimination of the methoxy group to yield the corresponding cyclic phosphate, is indicative of a possible mechanism of this transformation. The transition state seems to emerge as a result of the phosphorus atom being attacked by the 2-hydroxyl group of glycerol. In addition to the incoming group, the methoxyl group also finds itself in the axial position. The reaction ends in departure of the latter group.
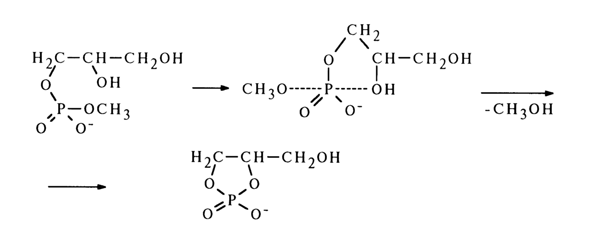
If the intermediate structure (17) had undergone pseudorotation [formation of structure (18)], the reaction products would have included glycero-2-methyl phosphate; the latter's absence indicates that no pseudorotation takes place in this case (this phenomenon will be discussed at greater length in what follows).
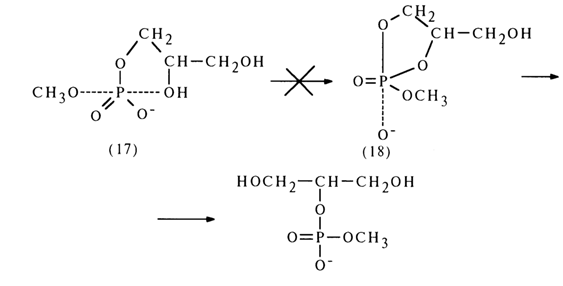
Migration of the phosphate group can occur in the case of non-alkylated glycol monophosphates:
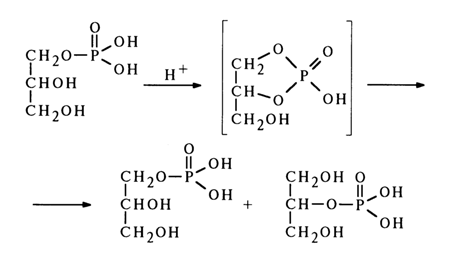
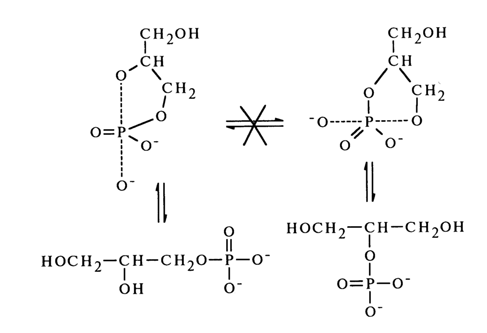
This process is possible only in an acid medium, which agrees well with the above-described mechanism of nucleophilic substitution at the tetrahedral phosphorus atom (it is the water molecule that is the leaving group in an acid medium).
Glycero-1 and glycero-2 phosphates are not capable of interconversion in alkaline media. Consequently, no cyclic phosphate is formed in this case. The reason is the lack of a suitable leaving group. It is most likely that although a cyclic structure similar to the one emerging in the transition state is formed, it is not capable of undergoing pseudorotations, which also rules out the possibility of isomerization.
Alkyl phosphates containing such electronegative substituents as C=-N, COOH, COOR and others at the b-position in the alkyl group readily undergo
b-elimination of the phosphate group in an alkaline medium. If ethyl phosphate practically does not break down during mild alkaline hydrolysis, b-cyanethyl phosphate and its esters do so easily under the same conditions.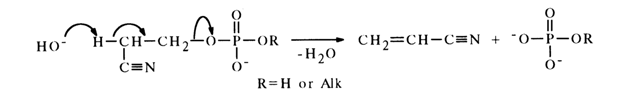
b
-Elimination of the phosphate group is always accompanied by cleavage of the C-O bond and formation of the corresponding unsaturated compound. The latter undergoes subsequent transformations in some instances. For example, during b-elimination of the phosphate group, serine phosphate is converted into pyruvic acid: