![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
11.5 Solid-Phase Synthesis of Oligodeoxyribonucleotides and Its Automation
Beginning from the mid sixties, the classical organic synthesis of oligonucleotides in solution evolved hand in hand with synthesis in a biphase system or, to be more precise, on insoluble polymer supports. This radical approach was for the first time proposed by Merryfield in 1962 for polypeptide synthesis.
Not long after the first results of its implementation as a means for building up a peptide chain had been obtained it became clear that it suits ideally oligonucleotide synthesis as well.
To begin with, no racemization can take place at the intermonomer node during formation of an internucleotide linkage (at least after removal of the protective group from the internucleotide phosphate in its synthesis by the triester method), and, consequently, there is no need to separate isomers after each monomer addition step. This drawback of solid-phase peptide synthesis at the early stages did not allow optically active polypeptides to be produced with good yields. Secondly, early in the evolution of oligonucleotide, as opposed to peptide, synthesis, long polymers were not needed during DNA synthesis. It was quite sufficient to synthesize 8- to 12-unit fragments to be used as building blocks for extended DNA duplexes. This is precisely why work on solid-phase synthesis of oligonucleotides began already when the phosphodiester method provided the only possibility to form internucleotide linkages - a rather ineffective procedure which did not allow each reaction of monomer addition to the emerging oligomer chain to be conducted with a good yield (exceeding 90%). In spite of the rather modest results, three university laboratories embarked on search for solutions to the many problems involved in oligonucleotide synthesis on polymer supports: in Evanstone (Letsinger), in Moscow (Shabarova) and in Heidelberg (Krahmer). The efforts of their research workers (Caruthers, Potapov, Köster, etc.) turned out to be consequential in the subsequent development of the most effective up-to-date method for automatic synthesis of oligo(poly)nucleotides. Later (in the mid seventies), this group of scientists was joined by representatives of the Cambridge school in England (Gait), where advances in the phosphotriester method were implemented within a short period of time. As far back as 1977, Gait published a seminal paper in NAR, describing the synthesis of 7- to 9-unit oligonucleotides some of which contained all the four DNA monomers, while synthesis of only 3- to 7-unit fragments, containing primarily the thymidine nucleotide, was reported by that time. The authors of the second paper on the subject, which made its appearance in NAR in 1979, were from Moscow University. They not only reported on synthesis of 10- to 15-unit nucleotides, including heterogeneous ones, but also presented the first map showing the operations involved in solid-phase synthesis and a schematic illustration of a semiautomatic synthesizer. The latter was a prototype of the synthesizer "Victoria" designed soon after in Novosibirsk. The next important event after these publications was the international symposium in Hamburg (Germany) held at Köster's initiative. It brought together all leading authorities on oligonucleotide synthesis. Many scientists with previous contributions to synthesis in solution spoke about synthesis on polymer supports. The culmination of the symposium was recognition of the solid-phase method as the most promising because it lends itself ideally to automation. Caruthers presented an important paper on highly effective application of the phosphite triester procedure to solid-phase synthesis. The years that followed were marked by extraordinary strides in solid-phase oligonucleotide synthesis and its automation.
11.5.1 Basic Principle of Solid-Phase Synthesis
The basic principle of the method is that synthesis is conducted in a two-phase system. One of the components (usually the nucleotide one) is covalently bound on a specially modified insoluble polymer and, consequently, is on the solid phase. The rest of the components are in solution. After condensation it is the oligonucleotide that is on the solid phase.
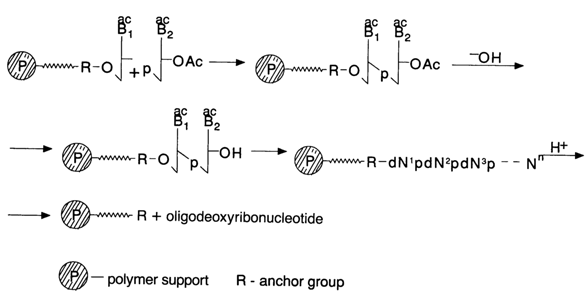
By simple filtration and washing it is separated from the reaction mixture and subjected to subsequent manipulations. Once the 3'-blocking group is removed, such a polymer-oligonucleotide is suitable for further condensation. As the chain continues to be built up, the target oligonucleotide is always on the solid phase from which it is separated only at the end of the synthesis. The complex oligonucleotide molecule is "assembled" in this manner, too.
What makes solid-phase synthesis of oligonucleotides particularly attractive is the drastic decrease in the time it takes, which is made possible by the extremely simple procedure of treating the reaction mixture and, what is most important. the fact that the reaction product does not have to be isolated after condensation (this is what takes most time). The simplicity of the procedure involving a polymer support makes it possible to make use of any excess reagents and also to repeat the treatment of immobilized molecules. Evidently, apart from taking much less time on the whole, the solid-phase method is the only one in which all operations can be standardized and the entire process automated.
The method, however, suffers from some shortcomings.
The steric hindrances inherent in the synthesis slow down somewhat the rates of all chemical reactions. The nonquantitative course of the reactions results in products with shorter chains accumulating on the polymer in addition to the target oligonucleotide. It is thus a rather serious problem to separate oligonucleotides after their removal from the polymer.
As suggested by the described principle of the solid-phase synthesis (we are considering it only in the context of oligonucleotides but it is equally applicable to any organic compounds), this process can be easily automated because it is essentially an iteration of three basic operations:
(1) covalent bonding;
(2) washing of the polymer;
(3) preparation of the immobilized molecule for the next step.
Elaboration of an effective procedure of solid-phase oligonucleotide synthesis, which would lend itself readily to automation, boils down to solution of at least two key problems:
(1) optimization of the support's macromolecular structure;
(2) selection of the right method to create the internucleotide linkage.
Solving the second problem was especially important because the entire synthesis hinged on it. If any of the synthesis steps is ineffective, the probability of obtaining an extended oligonucleotide is extremely low. For example, the yield in each step being 70 %, the overall yield of a hexanucleotide will be 12 %. At an 80 % yield, one can obtain a decanucleotide with the same yield. And if the yield during the formation of each internucleotide bond is 90 %, a 20-unit polynucleotide can be obtained with the same yield. In other words, solid-phase synthesis of oligonucleotides calls for condensation procedures with yields of at least 90 %.
11.5.2 Polymer Supports and Immobilization of the First Monomer Thereon
For successful outcome of reactions involving a polymer support, the latter must meet some general requirements. It must not influence the course of the synthesis. This requirement is practically hard to meet because condensations occur within the polymer which acts as some kind of a solvent and whose interactions with the immobilized nucleotide component and molecules in solution materially affect their properties. The support must exhibit minimal adsorptivity to ensure rapid and quantitative washing of the polymer to get rid of excess reagents and solvents; it must also be large enough to permit free diffusion of the reagents. At the same time, it must be mechanically and chemically stable as well as "compatible" with the solvents in which the synthesis takes place (solvatable) and also with the growing oligonucleotide chain. Another important consideration is how quantitative all reactions involving immobilized molecules (condensation, removal of the protective groups) are.
Practical introduction of polymer supports into oligonucleotide synthesis must be credited to Letsinger who did it in the mid sixties. Before the seventies, polystyrene-type supports were employed in all experiments. These are readily available polymers carrying aromatic groups and can be very easily modified through introduction, usually rather simple, of an anchor group (to which the first nucleotide unit is attached). Such polymers have a long shelf life and undergo no changes in the course of oligonucleotide synthesis.
In the early seventies, V K. Potapov proposed a new type of support for oligonucleotide synthesis, in which the best properties of previously studied polymer matrices were combined. The support was produced by chemical modification of polystyrene grafted onto the surface of polytetrafluoroethylene under radiation. The amount of grafted polystyrene can be easily controlled within 10 to 50 % of the teflon weight.
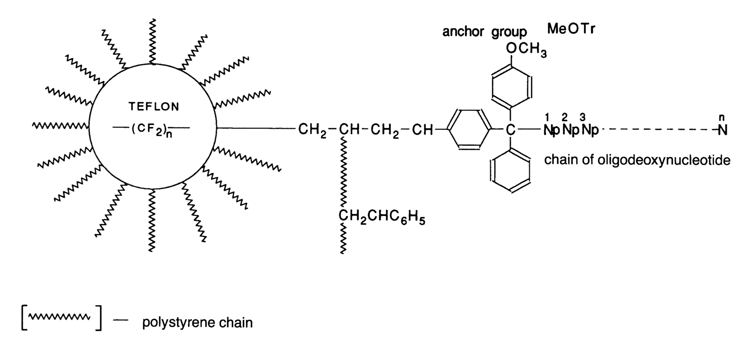
Transferring the reaction sites to the bead surface minimizes the role of diffusion processes, whereas the high conformational mobility of polystyrene chains not joined by divinylbenzene cross-linking mitigates steric hindrances both during modification of the polymer and in the course of oligonucleotide synthesis. The support displays low adsorption activity and swelling because the teflon matrix is totally inert with respect to all kinds of reagents and solvents. An outstanding feature of grafted supports, setting them apart from any cross-linked polymers, is the possibility of a high degree of modification (30-50%) by introducing anchor groups into such a support. Already in 1975-78, heterogeneous 8- to 10-unit oligonueleotides were obtained on this polymer support. Another support with a similar performance in oligonucleotide synthesis was polydimethylacrylamide proposed by Gait, which is embedded in macroporous kieselguhr. Just as the two supports mentioned earlier, this polymer may be quite useful in preparative synthesis of oligonucleotides. Yet another support, cellulose, is employed, similarly to polystyrene grafted to teflon and polyacrylamide/kieselguhr, only in the laboratories of origin. However, cellulose is used in the method of simultaneous synthesis on disks (see below). In the early eighties there appeared new types of supports having similar properties but more readily available, which accounts for the wide popularity they have gained in oligonucleotide synthesis. The main two types of supports are:
Controlled Pore Glass. This support is used more often than anything else for producing microamounts of oligonucleotides in automatic synthesizers. The size of pores in such a glass may be 240, 500, 1400 or 3000 Å. Glass with 500 Å
pores is most frequently used. Aminopropyl is employed as the "anchor" group (a group mediating between the polymer and the first nucleotide molecule from which the oligonucleotide chain starts to grow).Silica Gel. This is another widely used support in automatic synthesis with aminopropyl performing the function of the anchor group.
The almost universally adopted procedure of building up an oligonucleotide chain in solid-phase synthesis is based on a scheme with the starting point at the 3' end of the oligonucleotide; in other words, the synthesis proceeds in the 3'-5' direction. The main reason for this choice is the experimentally established fact that the rate of the reaction between the 3'-phosphate of the nucleotide component and 5'-hydroxyl of the nucleoside component is four times that between the 5'-phosphate and 3'-hydroxyl. This is why the first nucleotide unit is immobilized on the polymer support via its 3'-hydroxyl. To this end, one must first of all prepare a succinate of 5-dimethoxytrityl nucleoside, in which the second carboxyl is activated (in the form of an activated ester). The following scheme illustrates the sequence of steps in the preparation of modified silica gel and immobilization of the first nucleoside on its surface.
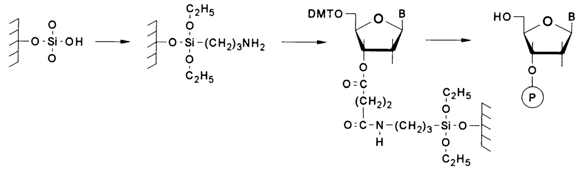
As a result of acylation of the amino group in the modified silica gel, the polymer and the first nucleotide unit are linked via an amide bond known for its high stability.
11.5.3 Phosphoramidite-Triester Method
Solid-phase synthesis of oligonucleotides has become much more effective since the early eighties, after radically new methods for forming internucleotide linkages were introduced, based on trivalent phosphorus compounds. The first to be introduced (see scheme below) was the so-called phosphoramidite method in which the nucleoside component is deoxynucleoside 3'-phosphoramidite.
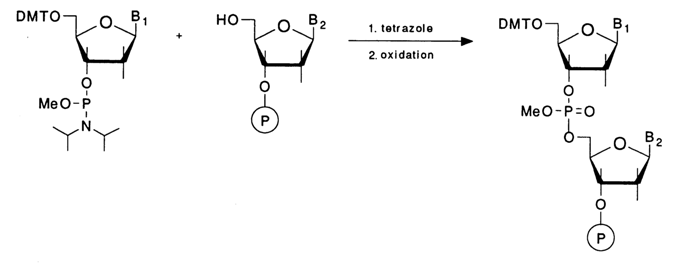
The phosphorylating function in the nucleotide component is performed by the trivalent phosphorus atom. Being intrinsically stable, phosphoramidites may enter into nucleophilic substitution reactions only after protonation. Caruthers, who elaborated this method for practical applications, proposed tetrazole (a very mild reagent and a proton donor) to be used for acid catalysis. The condensation proceeds almost quantitatively within 30 to 40 seconds. The next step is oxidation of the internucleotide phosphite group or, in other words, conversion of the trivalent phosphorus into a pentavalent one, which is also very fast (1-2 min) and quantitative in the presence of iodine and water. Significantly, the end product of such a two-step process (condensation, oxidation) is phosphotriester - that is, a compound structurally similar to the internucleotide phosphate forming during synthesis by the triester method. The protective group at the phosphate is a methyl easily and quantitatively removable by thiophenol. Notably, by virtue of the fast reaction rates this scheme can be used with any success only in solid-phase synthesis of oligonucleotides.
The following scheme with abridged formulas illustrates a single reaction cycle whereby a monomer becomes coupled to the polymer chain. Repetition of such cycles leads to extension of the oligonucleotide chain.
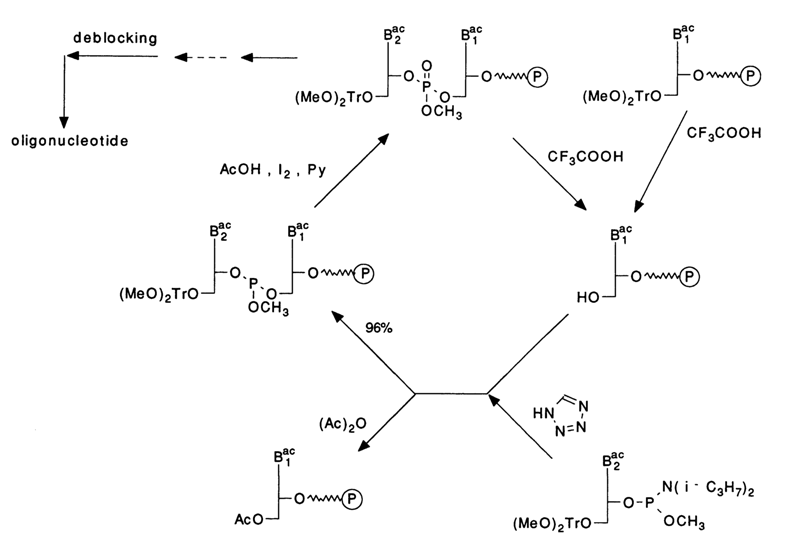
The first step of the cycle is removal of the (MeO)2Tr group from the 5'-hydroxyl (here, from the nucleoside coupled to the polymer support). To accomplish this, the polymer is treated with trichloroacetic acid (very mild conditions, not exceeding 1 min). After washing, protected nucleoside phosphoramidite and tetrazole are coupled to the support. The protonated amino group departs easily, and the phosphorus atom becomes sensitive to nucleophilic attack. As a result, a second monomer in phosphite form is coupled to the 5'-hydroxyl of the nucleoside associated with the polymer support. At this stage, the internucleotide phosphorus is still trivalent.
It may happen that the coupling reaction will not be quantitative (96-98 %). Some molecules of the nucleoside component coupled to the support may stay out of a reaction with phosphite. If they are left unreacted, their 5'-hydroxyls may enter into the reaction during the next step of coupling. This will give rise to an oligomer shortened by a unit (and so in each cycle), which will make it difficult to separate the mixture after synthesis. In order to rule out such impurities, the unreacted nucleoside component is blocked, or "capped". This is done by acetylation with acetic anhydride in the presence of N-methylimidazole and 2,6-lutidine. The 5'-acylated nucleoside component coupled to the support is now eliminated from the synthesis and remains uninvolved all the way until the oligo(poly)nucleotide chain is fully extended. After dimethoxytritylnucleoside phosphoramidite has been coupled, the phosphorus is trivalent. Therefore, the last step is oxidation, for which purpose iodine and water are used in the presence of a base (lutidine).
These five steps constitute a chain extension cycle which keeps being repeated.
Thus, the method resides in sequential extension of an oligonucleotide chain by using a respective activated monomer. The complete cycle takes three to seven minutes.
Once synthesis is over, the protective methyl groups are removed from the internucleotide phosphates (thiophenol), the oligonucleotide is removed from silica gel (aqueous ammonia, 200 C, 20 min), and, finally, the heterocycles are deprotected (concentrated ammonia, 600 C, 24 h). The dimethoxytrityl group blocking the 5'-hydroxyl serves several purposes:
(1) blocking of the 5'-hydroxyl throughout the reaction cycle when a nucleotide is coupled;
(2) determination of the yield of each step after decoupling of the (MeO)2Tr cation whose characteristic absorption occurs at 490 nm;
(3) as a lipophilic group during chromatographic separation of the reaction products (the target product is the only one having a (MeO)2Tr group at the end of the synthesis). This is especially important in the synthesis of extended oligonucleotides (with 20 and more units).
The advantages of the phosphoramidite method include the following:
(1) high yields in every step (96-98 %), which allow, firstly, the oligonucleotide chain to be extended by adding monomers, and that means easier preparation of the starting materials (there is no need to synthesize dimers or trimers in advance), and, secondly, the synthesis to be conducted at the micromole scale and purified 30- to 40-unit oligonucleotides to be obtained in an amount of 0.1 to 0.2 mg;
(2) lack of hygroscopicity of diisopropylamides of the protected nucleoside phosphates (there is no need to dry them prior to synthesis) and their stability in an anhydrous solution for two and weeks more, which allows the nucleotide component to be used in an absolute organic solvent over a long period of time;
(3) it takes only five to ten minutes for an internucleotide linkage formation cycle to be completed in the synthesizer.
The above scheme of extending an oligo(poly)nucleotide chain facilitates purification of the desired product. The post-condensation capping of the unreacted 5'-hydroxyl of the nucleoside component precludes growth of defective chains. It is only the target product that has a dimethoxytrityl group at the end of its chain, which makes its separation by reversed-phase high-performance liquid chromatography extremely easy. Detritylation (80 % acetic acid, 200 C, 2 min) yields a rather pure oligonucleotide. This procedure for building up oligonucleotide chains has become routine in laboratories equipped with automatic synthesizers.
11.5.4 Hydrophosphoryl Method
The other method based on trivalent phosphorus compounds is the hydrophosphoryl one shown schematically below:
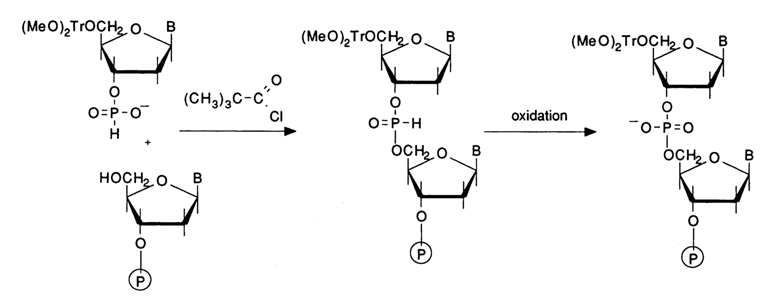
The phosphate in the nucleotide component is activated with the aid of such condensation agents as triisopropylbenzenesulfonyl chloride or tetrazole and N-methylimizadole. Experience has shown, however, that the best agent is pivaloyl chloride (PvCl) which brings the condensation yield up to 97-100%. The oxidation in the next step is performed with iodine in the presence of water. It has been found that dialkylphosphonates are oxidized with halogens during base catalysis. The general equation of the reaction involving iodine takes the form:
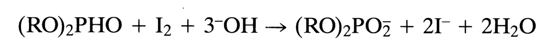
It has been demonstrated that the oxidation of the internucleotide phosphate group with iodine proceeds faster in the presence of N-methylimidazole, N-methylmorpholine, or triethylamine. The most universally accepted procedure involves a 2 % iodine solution in a pyridine-water (98:2) system. Here is a scheme with abridged formulas illustrating a reaction cycle in which a monomer is coupled to a nucleoside immobilized on a polymer support:
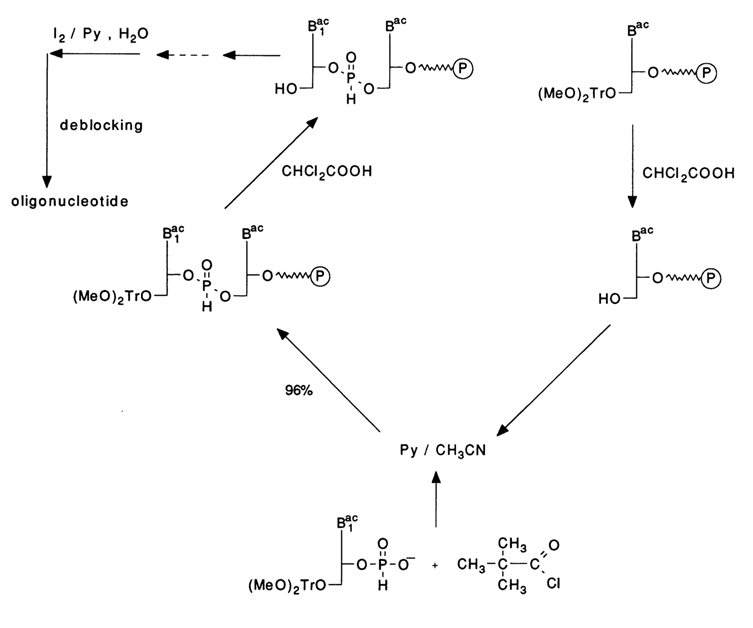
In the first step of this cycle, five aliquots of the phosphonate component and an aliquot of the 5'-hydroxyl nucleoside component condense in the presence of 50 mM PvCI in an anhydrous pyridine-acetonitrile (1: 1) mixture for five minutes. After preliminary detritylation of the nucleoside component in the presence of 2.5 % dichloroacetic acid in methylene chloride, the next condensation is carried out for two minutes. Then, after repeating the required number of cycles, the next step is oxidation with the following mixture: (a) 0.1 M I2 in pyridine/N-methylimidazole/H2O/tetrahydrofurane (5/1/5/90) or (b) 0.1 M I2 in triethylamine/H2O/tetrahydrofurane (5/5/90). In either case, the oxidation is over within two and a half minutes. Removal of the oligonucleotide from the polymer and protective groups from the heterocycle is achieved by treatment with concentrated ammonia (5 h, 550 C). The main difference between the hydrophosphoryl method of solid-phase synthesis of oligonucleotides and the above-described phosphoramidite one is the absence of a protective group at the internucleotide phosphate in the product of condensation and subsequent oxidation. Moreover, the reactions of the cycle proceed with 98 to 100 % yields. This allows the cycle to be shortened by several steps, and the absence of the step in which the protective group is removed from the internucleotide phosphate lowers the probability of side processes and the presence of lingering methyl groups in the sugar-phosphate backbone. All this has made the hydrophosphoryl method a procedure of choice for automatic synthesis of oligodeoxyribonucleotides.
The starting hydrophosphoryl derivatives of protected deoxynucleosides are synthesized by treatment of the corresponding N-acylated 5'-dimethoxytrityl nucleosides with a mixture of phosphorus trichloride, 1,2,4-triazole and N-methylmorpholine in anhydrous methylene chloride. The resulting hydrophosphoryl derivatives are rather stable and can be stored for a long period of time at room temperature.
11.5.5 Phosphotriester Method
The above-described phosphotriester method of oligonucleotide synthesis, based on intramolecular catalysis, has also proved successful with polymer supports:
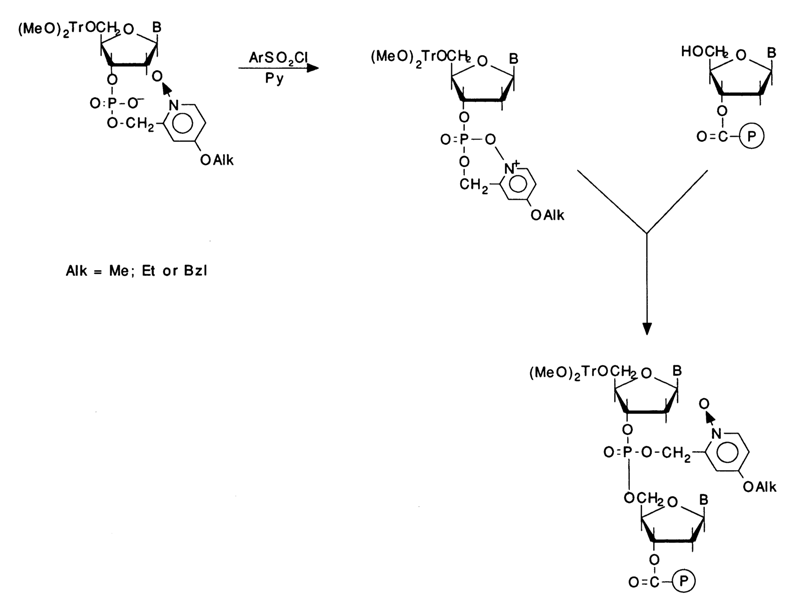
In this case, the fastest rate (30-60 s) of phosphotriester bond formation is ensured by using 1-oxide-4-alkoxy-2-picolyl derivatives in the presence of TPS. The yield of each condensation step is 98 to 99 %. The duration of one cycle is six minutes - that is, it is the same as in the phosphoramidite method.
The phosphate-protecting groups are removed from the synthesized oligonucleotides by treatment with piperidine. Notably, the synthesis can be performed virtually on any type of modern synthesizers, and the requirements of solvent drying are not as stringent as in the phosphoramidite case.
11.5.6 Automation of Synthesis
The standard nature of operations in the solid-phase method has led to automation of chemical oligonucleotide synthesis. The following diagram illustrates a general approach used in automatic synthesizer design.
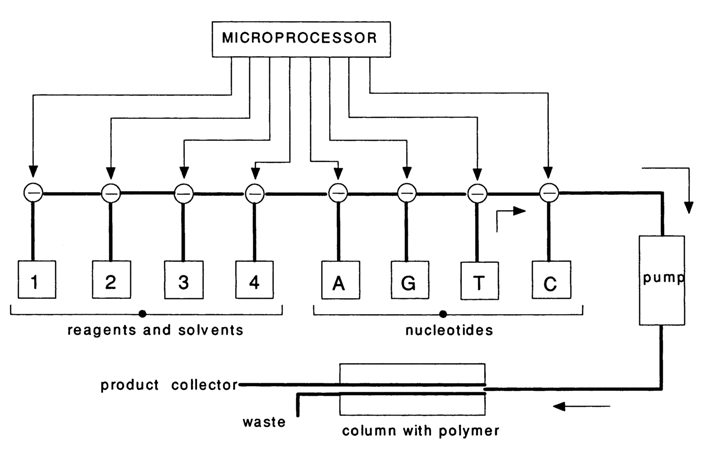
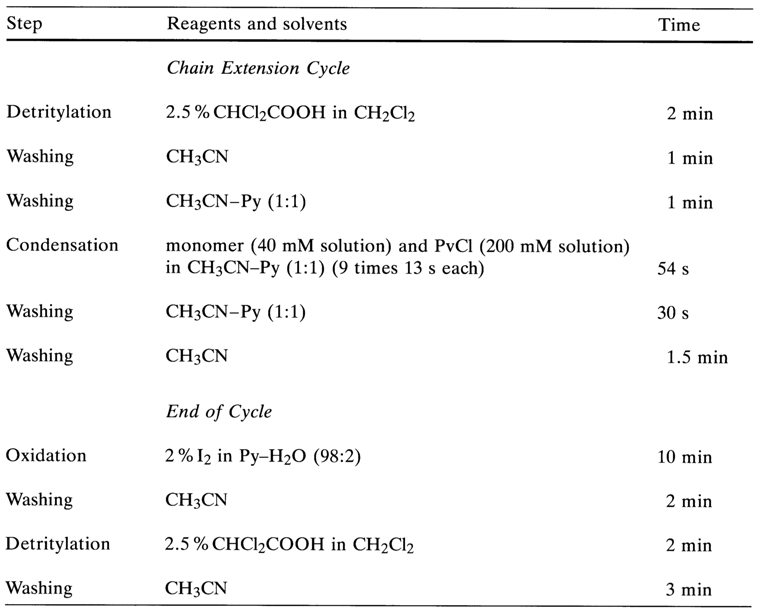
The operating principle of such a synthesizer is as follows. A microprocessor controls the pumping of protected nucleotide components (A, G, C and T), reagents, and solvents (1-4). All reagents are successively fed to a column containing a polymer support with the first nucleoside immobilized thereon. The synthesis and separation of the fully protected oligonucleotide from the polymer support are followed by deprotection, purification and analysis of the synthesized DNA fragments. Table 11-2 presents, by way of example, the sequence of operations to be performed in a synthesizer by the hydrophosphoryl method. The times listed in the table can be shortened. For some synthesizers a cycle takes three to five minutes.
The first Soviet synthesizer "Victoria" was developed already in 1980. At present, synthesizers "Victoria-5M" are produced by a special electronics and analytical instrumentation bureau under the USSR Academy of Sciences. In 1981-85, what are considered to be the best synthesizers in the West were
Table 11-2. Solid-phase Synthesis of Oligodeoxyribonucleotides by the Hydrophosphoryl Method.
purchased from such companies as 'Applied Biosystems", "Biosearch" and, later, "Pharmacia". A number of other companies also supply synthesizers.
Some models permit simultaneous synthesis of three or four oligonucleotides. Automation allows 30- to 40-unit oligodeoxyribonucleotides to be synthesized within just a few hours.
11.5.7 Multiple Simultaneous Synthesis on Polymer Segments
An alternative promising approach to solid-phase oligonucleotide synthesis involves polymer segments or, more specifically, cellulose (paper) disks. This procedure, sometimes also referred to as segment method, makes it possible to obtain a great number of oligonucleotides at a time. In this case, an oligonucleotide with a predetermined primary structure is synthesized not throughout the entire volume of insoluble polymer beads but on a small piece (segment) of cellulose or another material. Such a segment is used to immobilize the first nucleoside by the same chemical procedures as in conventional solid-phase synthesis. Elongation of the oligonucleotide chain does not necessarily have to involve just one segment. It becomes possible to attach the next monomer (one out of four) to several immolized nucleosides. One can use several segments with coinciding first and second nucleotides from the 3' end of the chain (the elongation proceeds in the 3'-5' direction, just as in conventional solid-phase synthesis). To perform the first two elongation steps, the disks are placed in columns isolated from atmospheric moisture, and the nucleotide component is passed through them under the usual conditions. Simultaneous synthesis of many oligonucleotides is conducted in four columns accommodating all segments, in which the chain starts to grow from the same monomer (i. e., there is coincidence with the structure of the second nucleotide from the 3' end). Each subsequent elongation step involves the same cycle. For this purpose, all segments intended for synthesis are marked with the numbers of the oligonucleotides to be synthesized, then four types of segments are prepared by immobilizing four nucleosides (the synthesis gives four types of oligonucleotides differing in the 3'-terminal nucleoside structure). After that, the segments are sorted out and placed in columns A, G, C and T, depending on the structure of the next nucleotide unit. Similarly, after the second step, the segments are sorted out and placed together in the same columns depending on the structure of the next unit. These operations are repeated as many times as necessary. This principle of simultaneous elongation of chains on six segments is illustrated below:
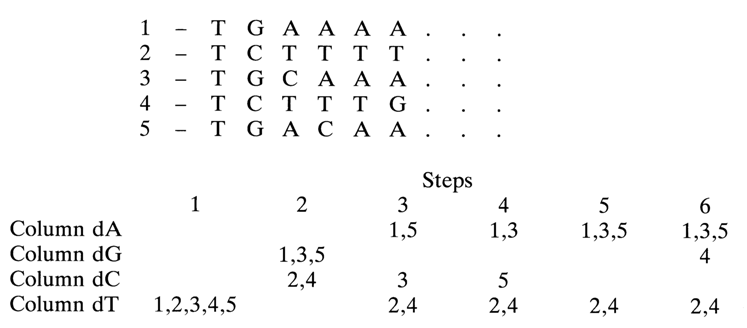
Synthesis is conducted simultaneously in this case - that is, all disks are involved in attachment of the next monomer (2nd, 3rd, 4th, etc.) at a time. The asynchronous elongation method has been proposed for synthesis of a great number of rather large (up to 30-32 b. p.) oligonucleotides. In this case, one can use a single column with the four nucleotide components being passed through it consecutively one at a time. To this end, an automatic one-column synthesizer is employed. The following example illustrates a plan for synthesizing ten fragments of two genes. g1 and g3, of melanocyte-stimulating hormones (g1-MSH and g3-MSH).

Given in column A is the primary structure of ten 12 to 34 b. p. oligonucleotides constituting both genes. Column B is a printout representing an optimal procedure for the simultaneous asynchronous elongation of each of the ten chains of the oligonucleotides listed in column A. It can be seen that a cytosine nucleoside is coupled to segments 4 and 10 during the first step (01), an adenine nucleoside is coupled to segments 2, 3, 7 and 10 during the second step (02), and so on. In other words, if dimers are formed at segments 2, 3, 4 and 7 during these two steps, we have already a trimer at segment 10. What makes the asynchronous elongation so attractive is the possibility to use a single-column synthesizer with all the advantages involved (automatic feed of the reagents, washing of the fluid, isolation from atmospheric moisture).
In the early work, the segments were of Whatman 3M or similar paper as well as mechanically stronger cellulose fabrics. The condensations were carried out by the usual triester method. Because of the relatively low yields (70-80 %), the condensation by this method yielded only 10 to 12 b. p. sequences. In recent years, Seliger's laboratory (Germany) introduced synthesis on Fractosil 500 support (the segments are prepared in a special way by the phosphoramidite-triester method). In this case, the yields at the elongation step average 95 % with the result that chains with up to 30-35 units can be produced.
There is every possibility that the triester method with a phosphate-protective group capable of O-nucleophilic catalysis will also be rather effective.
11.5.8 Separation and Purification of Synthetic Oligonucleotides
Liquid chromatography is the basic method for separating and purifying oligonucleotides. In the phosphodiester case, oligonucleotides undergo ionexchange chromatography. If synthesis is done by the triester method, partition chromatography on silica gel is used. Separation of oligonucleotides with protected internucleotide phosphates is often accomplished by reversed-phase chromatography. This approach is especially useful if the oligonucleotide contains a 5'-dimethoxytrityl group. In this case, it is easily separated from less lipophilic oligonucleotides lacking such a group on silica gel C-18. High-performance liquid chromatography (HPLC) on silica gel C-18 is also recommended for the same purpose. This chromatographic technique is instrumental in analysis and fractionation of deprotected oligonucleotides as well as separation and purification of compounds produced by solid-phase synthesis. Oligonucleotides separated by reversed-phase chromatography must be assayed by ion-exchange chromatography or gel electrophoresis for homogeneity (separation by charge or chain length). Another method for identification and purification of synthetic oligonucleotides is electrophoresis in polyacrylamide gel. This technique provides for separation by charge and allows rather extended (up to 200 b. p.) sequences to be obtained. Among its drawbacks is that it is impossible to separate preparative amounts of oligonucleotides. Moreover, quantitative separation from the gel is also impossible. PAGE is especially good for separation and purification of oligonucleotides synthesized on polymer segments. It is conducted under denaturating conditions (to avoid complexing) in 7 M urea or in the presence of formamide.