![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
11.4.2 Chemical Synthesis of Oligodeoxyribonucleotides in Solution
11.4.2.1 Phosphodiester MethodThis method was the first to be used for synthesis of oligo- and polydeoxynucleotides and, therefore, has been studied most thoroughly. Most of the credit for its development, including preparation of the nucleotide and nucleoside components, activation of the phosphate group with the aid of DCC and Ar2SO2C1 (see above), as well as elaboration of procedures for elongating the nucleotide chain, must be given to Khorana and coworkers.
Synthesis of oligodeoxyribonucleotides with a chain 8 to 12 monomer units long by Khorana's method includes, at the early stages, condensation of the corresponding (5'-terminal) deoxyribonucleoside (13), in which the 5'-hydroxyl is blocked by a para-methoxytrityl group, together with the nucleotide (14) in which the 3'-hydroxyl is protected by an acetyl group. The condensation is performed in the presence of 1,3,5-triisopropylphenylsulfonyl or mesitylenesulfonyl chlorides:

After removal of the 3'-acetyl group, the resulting protected dinucleoside phosphate (15) is subjected to alkaline treatment to be converted into the nucleoside component for subsequent elongation of the oligonucleotide chain. Treatment of the dinucleoside phosphate (15) with ammonia (to eliminate the acetyl and protective groups from the heterocyclic bases) and dilute acid (to eliminate the monomethoxytrityl group) gives a deprotected dinucleoside phosphate.
The condensation is carried out in absolute pyridine which, as has already been pointed out, is indispensable for activating the nucleotide and also capable of dissolving even high-molecular weight oligonucleotides (in the form of the corresponding derivatives). Another advantage of this solvent is its relatively high volatility, which makes its removal from the reaction mixture possible under very mild conditions.
The resulting partially protected oligonucleotide (16) is purified by extraction with organic solvents, which is quite possible because it contains a lipophilic trityl group, or by ion-exchange chromatography on DEAE cellulose.
Subsequent elongation of the oligonucleotide chain is possible by stepwise addition of monomers of type (14). This is what is known as the step-by-step method. It is generally used to obtain three- to four-unit oligonucleotides. Step-by-step synthesis of longer oligonucleotides creates difficulties in separation. The reason is that every new unit added to the nucleotide chain in such synthesis leads to smaller charge and molecular weight increments. This is precisely why the block method had to be adopted. It resides in attachment to the nucleoside component (16) of a nucleotide one which is essentially a di- or trinucleotide (at later stages this component may comprise even a greater number of monomer units). Synthesis of the latter may proceed as follows, for example:
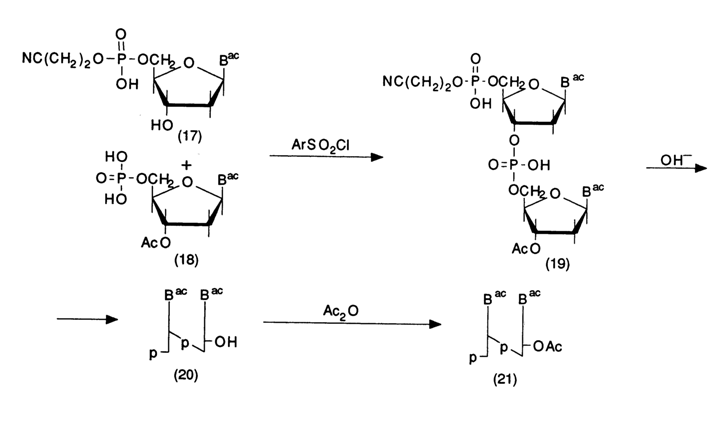
The condensation is conducted as described above; it should be noted here that the nucleoside component (17) is also a nucleotide. Its difference from the nucleotide component (18) is that its phosphate group is blocked by cyanoethyl. After condensation, the protected dinucleotide (19) is treated with an alkali (both the cyanoethyl and acetyl or acyl groups are removed) then, after chromatography on DEAE cellulose, with acetic anhydride which selectively acetylates the 3'-hydroxyl group of the dinucleotide (20). The emerging partially protected dinucleotide (21) is used for the elongation of the oligonucleotide chain block by block. This approach was implemented in Khorana's laboratory for preparing large quantities of 8- to 12-unit oligodeoxyribonucleotides from which the complete tRNATyr gene was then constructed. The following scheme illustrates the synthesis of undecadeoxyribonucleotide, which is one of the fragments if this gene.
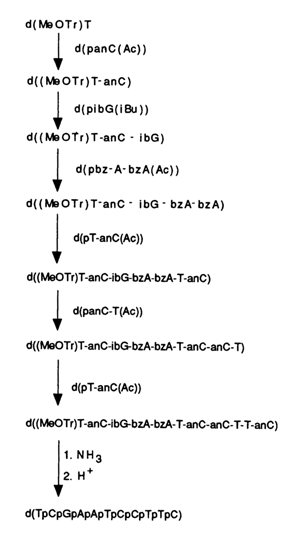
What sets this synthesis path apart is the fact that the 5' end of the chain always retains an alkali-stable para-methoxytrityl group, while the 3' end contains, after each step of synthesis, an acetyl group which is selectively eliminated by alkaline treatment prior to the next step (see Table 11-1). Thus, the chain is built up from the 5' to the 3' end. When the synthesis is over, sequential treatment with ammonia and a dilute acid removes all protective groups, and the oligonucleotide is carefully purified and analyzed by ion-exchange chromatography. This scheme has been successfully used in the synthesis of many biologically active 8- to 12-unit oligodeoxyribonucleotides.
If polydeoxyribonucleotides containing more than 12 monomer units are to be synthesized, the effectiveness of the block method also goes down for two reasons. Firstly, the yields at every subsequent step of condensation drop drastically. This decrease in yields stems from side processes, primarily those due to cleavage of internucleotide linkages via intermediates of triester type (7) and also, in all likelihood, steric hindrances to the condensation.
When oligonucleotides containing more than 10 to 12 units are joined, the yields of condensation products become extremely low and cannot be raised even if the nucleotide component is used in markedly excess amounts (up to 200-fold). The steric hindrances to condensation during synthesis of polynucleotides longer than 10 units are most likely due to formation of intermediate compounds of type (6) (products of addition of an active nucleotide component to every internucleotide phosphate group). The result may be a globule in which the 3'-hydroxyl end of the polynucleotide may be drawn into the molecule by virtue of non-covalent interactions (e. g., hydrogen bonding). The other reason for low effectiveness of block-by-block elongation of the chain has to do with difficulties in purification and separation of the desired polynucleotide by ion-exchange chromatography.
The above method has been used for one-step synthesis of oligo- and polydeoxyribonucleotides by way of polycondensation of appropriately protected mono- and oligonucleotides. In the simplest case, if only the heterocyclic base is blocked in the nucleotide, homogeneous oligo- and polynucleotides are formed in the presence of a condensation agent (the apostrophe indicates the presence of a protective group in the base residues):
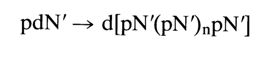
A similar course is taken by the polycondensation of oligonucleotide segments in which only heterocyclic bases are protected:
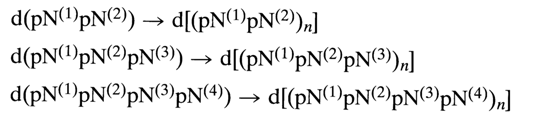
This is how homogeneous oligonucleotides or oligo- and polynucleotides
with alternating di-, tri- and tetranucleotide sequences can be obtained at a sufficiently
fast rate. A drawback of the method is the possibility of many side processes. In addition
to those already described, we may have formation of cyclic oligonucleotides ![]() and various pyrophosphates as a result of
intramolecular phosphorylation. Serious difficulties arise during separation of oligo- and
polynucleotides as well as their identification. None the less, such synthetic oligo- and
polynucleotides with alternating di-, tri- and tetranucleotide segments became available
in the mid sixties and played a key role in establishing the structure of the amino acid
code. Homo- and heterogeneous polycondensation products continue to be extremely important
research tools in molecular biology.
and various pyrophosphates as a result of
intramolecular phosphorylation. Serious difficulties arise during separation of oligo- and
polynucleotides as well as their identification. None the less, such synthetic oligo- and
polynucleotides with alternating di-, tri- and tetranucleotide segments became available
in the mid sixties and played a key role in establishing the structure of the amino acid
code. Homo- and heterogeneous polycondensation products continue to be extremely important
research tools in molecular biology.
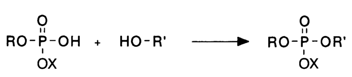
The above-mentioned drawbacks of the phosphodiester method have been obviated to a large extent by blocking the phosphate group prior to synthesis of the internucleotide linkage; as a result, the emerging intermonomer linkage has a phosphotriester structure:
It took quite some time for the method to gain popularity, although, historically, this is precisely how the first oligonucleotide was obtained by Michelson and Todd. There are many reasons for this lack of popularity till 1973. First of all, it was the fact that no condensation agent capable of activating the diphosphate as effectively as a monophosphate was available.
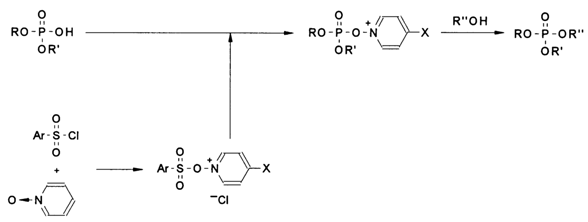
It was also important to find suitable protective groups to block the internucleotide phosphate. In 1965, Letsinger and coworkers came up with a proposal to use 2-cyanoethyl, which can be easily eliminated by mild alkaline treatment (
b-elimination), as the protective group and TPS as a condensation agent. In 1967, cyanoethyl was replaced by trichloroethyl which can be quantitatively eliminated by treatment with zinc powder in 80 % acetic acid, and the following year it was superseded by an alkali-labile phenyl group. None of the above groups, however, could meet all the requirements of oligonucleotide synthesis (ease of incorporation, stability during synthesis, ease of removal). In some cases, by-products with internucleotide (3'-3' and 5'-5) bonds not existing in nature were formed, in others, the internucleotide bonds were ruptured in a most pronounced manner. Nevertheless, it was at that stage that the basic principles of the general strategy for building up oligonucleotide chains by the triester method were formulated. It became obvious that deoxyribonucleosides were the starting compounds for synthesis and that the best way to prepare the nucleotide component is through phosphorylation of the 5'-protected nucleoside. However, the continuing application of the proven approach (diester method) to elongate oligonucleotide chains and use of arylsulfonyl chlorides slowed down implementation of the triester method. A major breakthrough for the latter took place in 1973-74 when a new type of arylsulfonic acid derivatives emerged as condensation agents. This is when the main advantage of imidazolides, namely, their much lower aggressivity (the reaction mixture becomes strongly resinous in the presence of TPS) was pointed out, although the internucleotide linkage formation proceeds at a slower rate. In 1974, Narang and coworkers described the highly successful use of triazolides of arylsulfonic acids in the triester synthesis. From that moment on, the triester method, which is much more effective than the diester one, started making deep inroads into oligonucleotide synthesis.Diphosphate activation techniques have been rapidly improving. Tetrazolides of arylsulfonic acids have turned out to be the best condensation agents.
In the meantime, modifications of the method appeared. For example, in 1982 V. A. Yefimov and coworkers proposed the use of ArSO2C1 in the presence of N-methylimidazole in neutral organic solvents (acetonitrile, nitromethane, etc.). In 1985, the same team introduced more powerful nucleophilic catalysts into the phosphotriester method, namely, N-oxides of 4-substituted pyridines in the combination with ArSO2C1, which enhances considerably the rate of phosphodiester bonding (1 min) and brings it closer to the rates typically observed in phosphite methods.
In Krahmer's laboratory there was proposed an integrated approach to synthesizing oligonucleotides by the triester method, which, in its general form after undergoing some changes, underlies the current strategy of synthesis by this method.
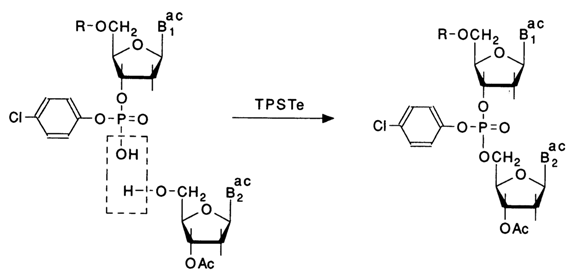
As has already been mentioned, a major contribution to this oligonucleotide synthesis method was made by Narang who further elaborated it and proposed a new group (substituted aryl), in addition to the new condensation agent, for blocking the internucleotide phosphate:
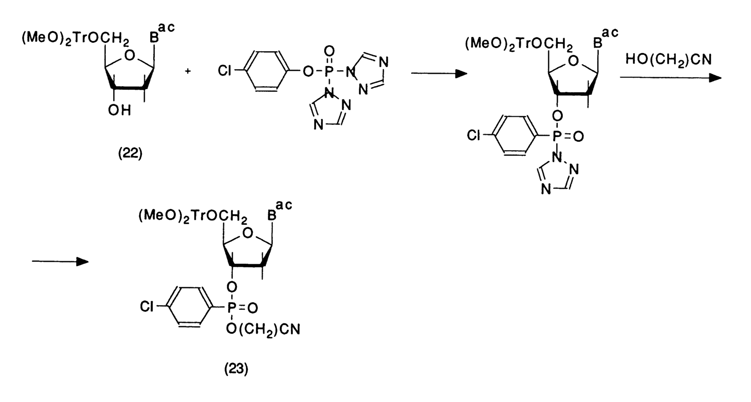
T'he key compounds in this scheme are deoxynucleosides with protected 5'-and 3'-hydroxyl groups. Deoxyribonucleoside (22) with a di(para-methoxy)trityl group at position 5' is phosphorylated with bis-triazolide of para-chlorophenylphosphoric acid, and fully protected nucleotide (23) is obtained after treatment with ethylene cyanohydrin. The phosphorylating agent is derived from para-chlorophenyl dichlorophosphate by treatment with two aliquots of 1,2,4-triazole in the presence of triethylamine. Because of its extraordinary lability it is not isolated but entered immediately into the reaction. The protected nucleotide (23) is a key compound and can serve as a source, after selective elimination of protective groups, of both nucleoside (24) and nucleotide (25) components:
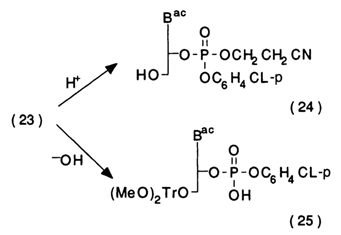
Compounds (24) and (25) are then entered into a reaction with a condensation agent, usually tetrazolide of arylsulfonic acid. The reaction is conducted in absolute pyridine for one or two days as follows:
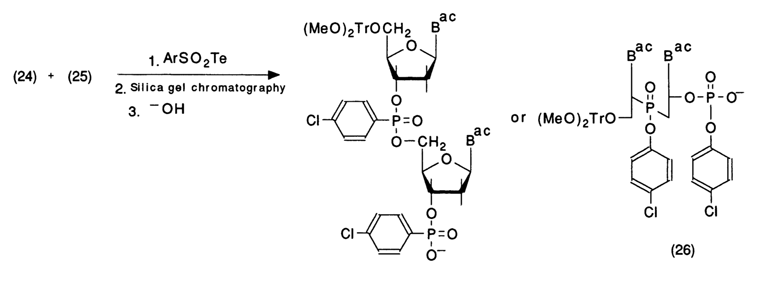
In this case, in contrast to the phosphodiester method, the condensation proceeds at a slower rate. This seems to be due to the steric hindrance to the reagents (condensation agent and nucleotide component) and (which may be the main reason) a different mechanism of substitution at the nucleotide phosphate. The active form of the phosphorylating agent here is tetrazolide (27) of the nucleotide component, which is formed immediately after elimination of tetrazole according to the scheme:
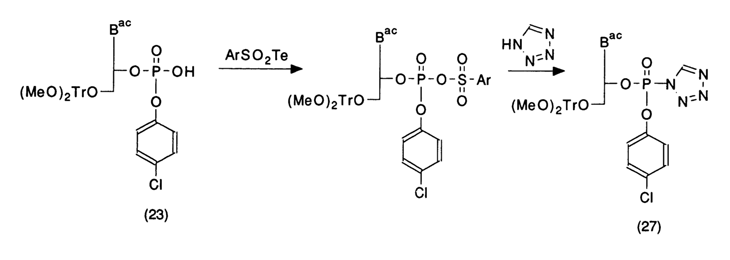
Compound (27) must be much less reactive than the pyridine-stabilized metaphosphate used as the condensation agent in the diester version of synthesis.
Oligonucleotide (26) may be further entered into the condensation reaction either with a nucleotide of type (24) to give oligonucleotides containing a phosphate group at the 3' end or with the protected nucleoside (29) to yield trinucleoside diphosphate (30) according to the following scheme:
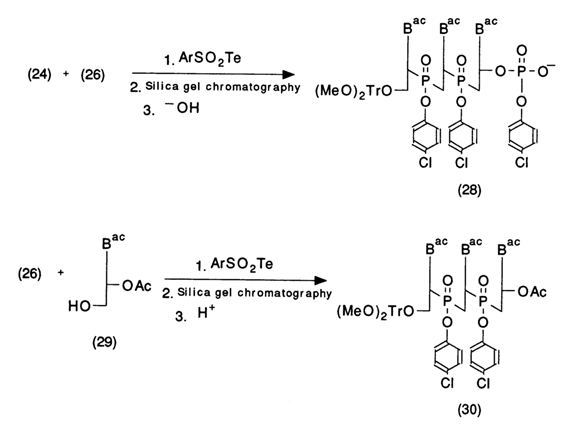
Condensation of (28) and (30) type compounds results in oligonucleotides with six monomer units which can be used in a similar fashion for further elongation of the oligonucleotide chain. Here is a standard scheme of the synthesis of 10- to 20-unit oligonucleotides.
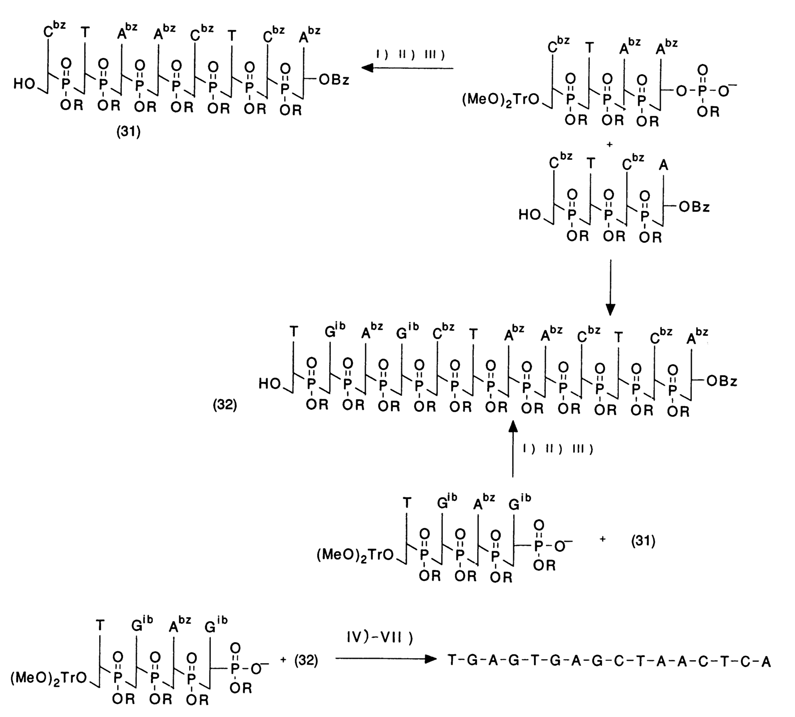
The protective group for the internucleotide phosphate is selected in such a way as to ensure its easy removal at the end of the synthesis. For selective removal of the acid-labile (MeO)2Tr group an oligonucleotide is treated with benzenesulfonic acid in chloroform at 00 C. Under such conditions, the depurination of a protected oligonucleotide is virtually nil.
A selective removal of the 2-cyanoethyl group in compounds of type (24) is achieved by treatment with 0.2 N sodium hydroxide in dioxane, when oligonucleotides are short, and with anhydrous triethylamine in pyridine, when they are long. All condensations are carried out using equivalent amounts of the nucleoside and nucleotide components in 2,3,5-triisopropylbenzenesulfotetrazolide (TPSTE) as the condensation agent. After each condensation, the oligonucleotide is separated by silica gel chromatography.
A comparison of the triester and diester methods reveals the most important advantages of the former: fewer side reactions, stability of yields in each step of the synthesis even when long oligonucleotides are formed, and ease of their purification by adsorption chromatography on silica gel.
Within a short period of time, however, the accumulated experience with the above-described triester synthesis procedure brought to light some of its drawbacks. The main problem, lowering the synthesis rate, has to do with the separation of fully protected oligonucleotide segments by silica gel chromatography, and the longer the chain, the worse the separation (this depends also on the nucleotide sequence). Itakura and coworkers proposed to eliminate the intermediate purification of oligonucleotide segments and, accordingly, altered slightly the condensation conditions. In order to bring the condensation to a quantitative level, the nucleotide component was entered into the reaction in a doubly excess amount. Notably, the entire synthesis was conducted by combining trinucleotide segments as shown below:
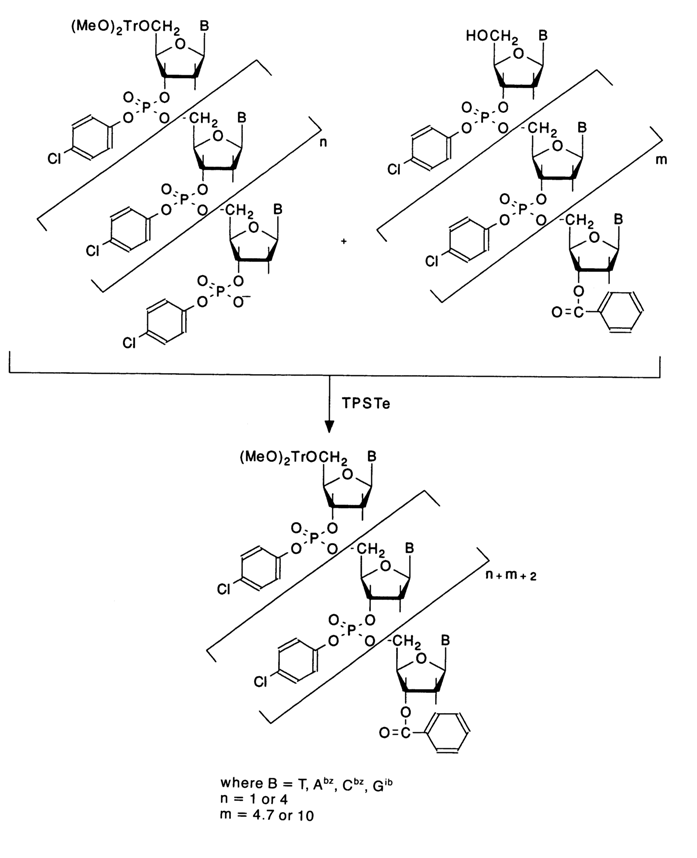
The excess of the nucleotide component was removed by treatment with an ion-exchange resin (DOWEX 1x8 or DEAE cellulose) in aqueous organic solvents, and a fully protected oligonucleotide was separated after chromatography on very wide (7 cm in diameter) and short (3 cm) columns. When this oligonucleotide was treated with a 2 % solution of benzosulfonic acid in a methanol-chloroform (3:7) mixture to remove the (MeO)2Tr group, the resulting one (nucleoside component) was used to further build up the chain without additional purification. Assembling nucleotides from trinucleotide segments makes it possible to easily separate the end product from the mixture of intermediate oligonucleotides at the last stage, after elimination of all protective groups, by liquid chromatography on Permaphase AAX. According to the authors, all these improvements allow one, when trinucleotide segments are available, to speed up several times the synthesis of oligodeoxyribonucleotides of a desired structure.
As was pointed out in the introduction to this section, new nucleophilic catalysts (N-methylimidazole, N-oxides of substituted pyridines) have enabled the phosphodiester method to be considerably improved and expand the spectrum of modern techniques based on trivalent phosphorus compounds.
When N-methylimidazole is used (in an amount twice that of arylsulfonyl chloride) in synthesis according to the scheme just described, the reaction takes 5 to 10 minutes in the case of short oligomers and 20 to 40 minutes in the case of 16- to 20-unit ones. The level of modification of the heterocyclic bases has gone way down, which makes purification of the target oligonucleotides easier.
Even more advanced is the procedure proposed by V. A. Yefimov and coworkers, whereby condensations are carried out in the presence of 0nucleophilic catalysts of the pyridine N-oxide type. The reaction rate was maximum when N-oxides of 4-dimethylamino- and 4-alkoxypyridines were used. The internucleotide condensation takes only a minute in the presence of arylsulfonyl chloride and such a catalyst - that is, it proceeds at a rate several times faster, as compared to N-methylimidazole. In spite of their low basicity (pKa 2-4), N-oxides of pyridines exert not only nucleophilic but also basic catalytic effect and act as acceptors of the acids evolving in the course of the reaction. This is why the latter can be conducted not only in pyridine but also in other organic solvents. Here is a possible course that the reaction may take:
A further improvement of this technique was using a protective phosphate group as a catalyst. Such a group speeds up substantially the internucleotide condensation in the presence of arylsulfonyl chlorides. The increase in the phosphodiester bonding rate is most likely due to formation of an active cyclic intermediate:
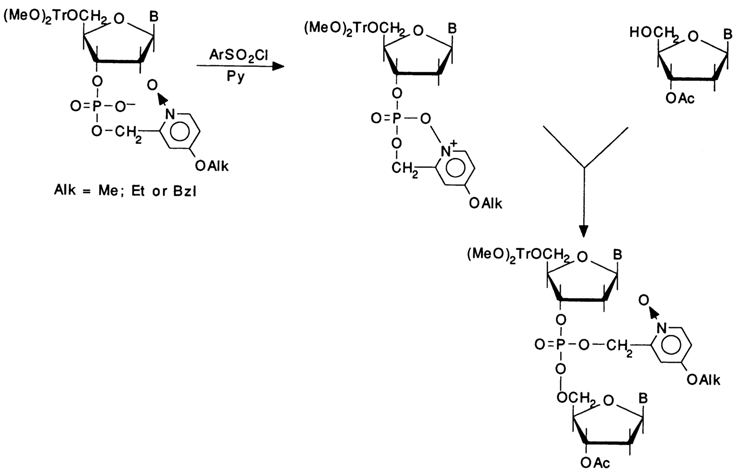
Condensation involving such intramolecular catalysts and ArSO2C1 is over within less than a minute. Studies into the stability of O-nucleophilic protective groups have shown that they are stable toward acid reagents used to remove the 5'-trityl groups and also during internucleotide condensations. They are eliminated, upon completion of oligonucleotide synthesis, in the presence of triethylammonium thiophenolate or piperidine. Advantages of this modification of the phosphotriester method include not only a very high speed of synthesis but also a very low level of side processes (5'-sulfonylation, heterocycle modification).
Monomers carrying catalytic phosphate-protective groups are prepared from the corresponding fully blocked nucleoside phosphotriesters by selective removal of the noncatalytic aryl or 2-cyanoethyl phosphate-protective group. Synthesis of the starting triester may take one of the following routes:
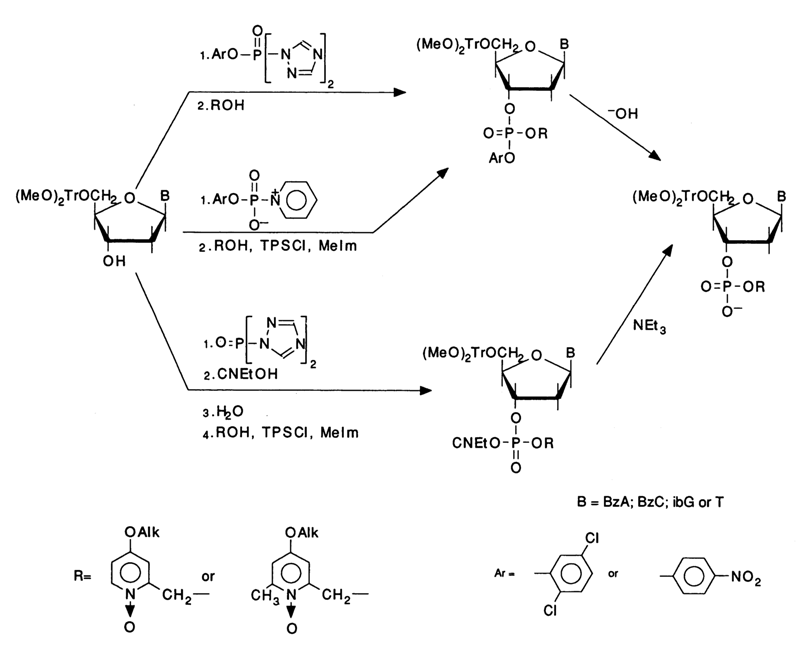
This particular modification of the phosphotriester method has been successfully used for automated solid-phase synthesis as well.
Now that simpler and more effective methods for creating internucleotide linkages with the aid of trivalent phosphorus derivatives are available, interest in the phosphotriester method has faded perceptibly. When the latter is used for synthesis in solution, however, its importance remains unabated for preparation of moderately long (15 to 20 b. p.) oligonucleotides, which is essential in developing new generations of oligonucleotide-based drugs.
In this section we have not touched upon the highly important aspects of individual steps of synthesis, such as selection of protective groups, their selective and complete removal, and the side processes accompanying each of these steps. Detailed and competent discussion of these matters can be found in Reese's review (see References).
11.4.2.3 Phosphite triester MethodIn 1975, Letsinger proposed a new method for creating internucleotide linkages. Its main difference from the previously described methods was the use of trivalent phosphorus derivatives which are highly effective phosphorylating agents. What makes this method especially attractive is the fact that phosphorylation of the hydroxyl group proceeds at a rather fast rate (1-2 min) and a very low temperature (-780 C):
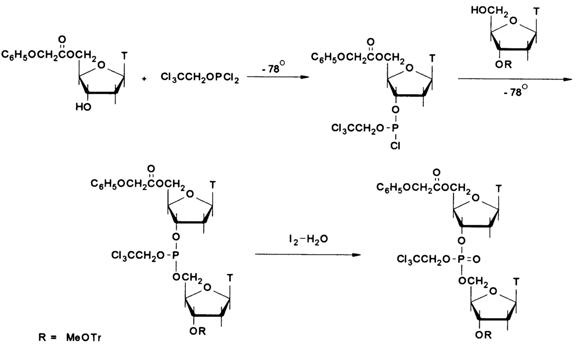
That first attempt had laid the foundation for the most fruitful approach in oligonucleotide synthesis, which became the most widely used method to synthesize oligo- and polynucleotides already in the early eighties.
As can be seen from the above scheme, the method includes the phosphorylation of a 5'-protected nucleoside with alkyl dichlorophosphate and another nucleoside with the resulting active derivative of the 5'-hydroxyl, followed by oxidation of the internucleotide phosphite group to the corresponding phosphate. Various alkyls and aryls were used at the early stages as protective groups in phosphates. The methyl group has turned out to be the most convenient by virtue of its easy removability after synthesis through treatment with trialkylammonium thiophenolate. Moreover, methylphosphite diazolides marked by greater stability started being used:
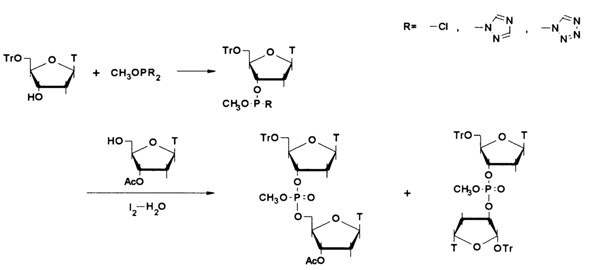
The most serious drawbacks of the processes illustrated above is the need to conduct the reactions at an extremely low temperature, as well as formation of by-products with an internucleotide 3'-3' bond. Besides, phosphite derivatives were found to be rather unstable and sensitive to even traces of moisture.
A major contribution to the evolution of the phosphite method for creating internucleotide linkages has been made by Caruthers. The di(isopropyl)amide or morpholide derivatives of phosphates proposed by him in the early eighties turned out to be much more convenient in use. They are characterized by moderate reactivity (reactions involving these compounds proceed effectively at room temperature), long shelf life even at room temperature, and lack of hygroscopicity.
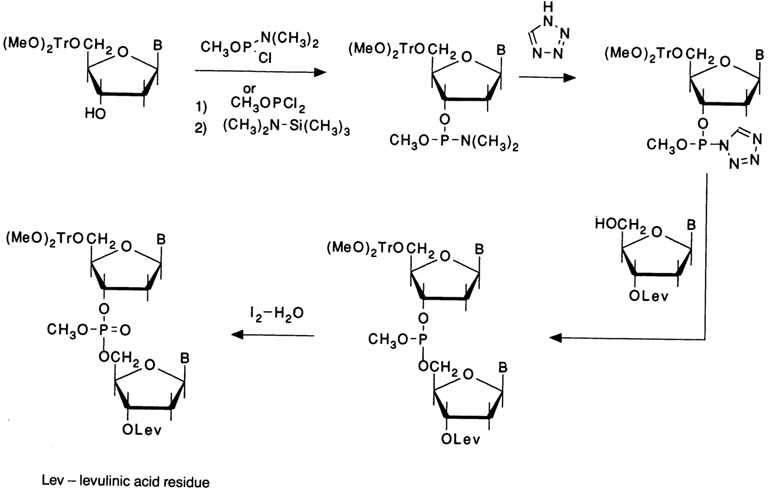
These and similar phosphorylating agents are employed most extensively in solid-phase synthesis of oligonucleotides.