![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
11.6 Synthetic Genes and Their Cloning
11.6.1 Methods of Duplex DNA Construction
Section 11.3 outlined the basic principle of gene formation with the aid of the enzyme T4 DNA ligase, and an example was given illustrating assembly of a tyrosine suppressor tRNA gene. The latter was assembled from synthetic oligonucleotides step by step. At first, short double-stranded fragments of types A, B and C, having sticky ends, were produced before assembling the complete gene.
An alternative approach involves direct ligation of all oligonucleotide fragments constituting a gene (one-pot ligation). To this end, a mixture of fragments is heated, cooled and then subjected to ligation. Some examples of such assembly are given in Table 11-3. Direct ligation is sometimes performed in the course of cloning; that is, the mixture of oligonucleotides with a linear vector is incorporated into competent cells already containing DNA ligase.
Recently, subcloning has become a widely used procedure. What it boils down to is preliminary cloning of intermediate DNA duplexes (with 60 to 100 b. p.) to be assembled into a gene. The intermediate double-stranded fragments must have sticky ends corresponding to a particular restriction endonuclease. This procedure allows rather pure fragments to be obtained. Their structure is determined by sequencing.
11.6.2 Synthesis of Genes and Their Functionally Important Segments
It has already been mentioned that the first chemoenzymatic synthesis of a complete tyrosine suppressor tRNA gene was performed in Khorana's laboratory. In addition to the structural gene of pre-tRNA, the synthesized 207 b.p.-long duplex also included a promoter (51 nucleotides) and a segment with a signal for processing the primary transcript of the gene (25 nucleotides) into functional tRNA. The synthesized double-stranded polynucleotide had sticky ends resulting from digestion of the corresponding sequence by restriction endonuclease EcoRI (AATT).
The gene was assembled, according to the scheme presented earlier, from duplexes I-V, P1-3 and P4-10.
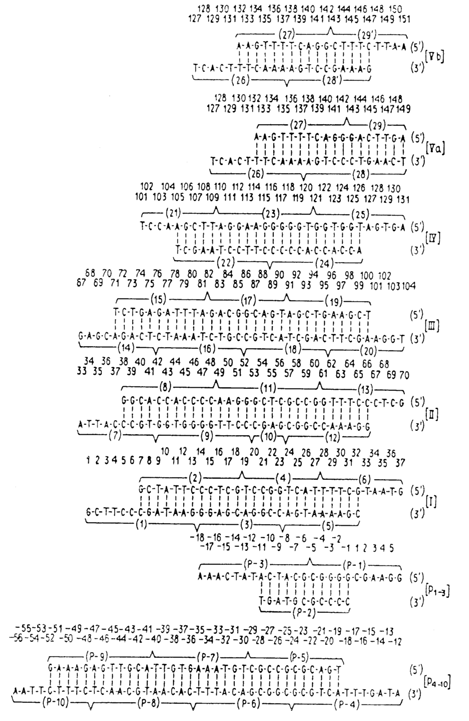
The scheme below illustrates the structure of the gene: symbols (_
?_) are used to indicate the sites at which the oligonucleotides are linked; also shown is the structure of the RNA-transcript obtained by transcription with a primer, namely, tetraribonucleotide C-C-C-G complementary to the gene fragment (-3) - (+ 1) (marked by symbol -).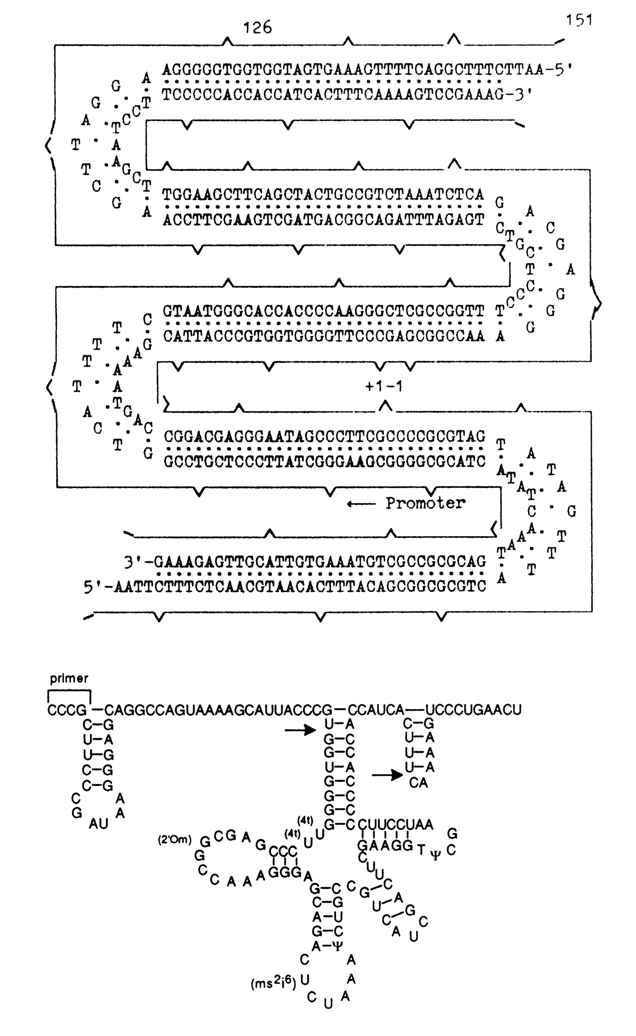
Analysis of the primary transcript has demonstrated that the nucleotide sequence of a gene whose chemical synthesis required 400 condensations was correct. Addition of an extract from E. coli S-100 to the primary transcript leads to its processing and modification. The primary transcript is digested with RNAse P to yield a 5'-terminal sequence (left arrow). During 3'-terminal processing (right arrow), the endonuclease breaks the linkage between the seventh and eighth nucleotides from CCA, then it sequentially detaches the seven "excess" nucleotides. Modification of the primary transcript is not consistent. The rT
yCG segment shows complete conversion of uridine into pseudouridine, but only 20 % of uridine are converted into ribosylthymine. In the anticodon loop, the rate of conversion of uridine into pseudouridine is 30%. Formation of 2'-O-methylguanosine and 2-methylthio-6-isopentenyladenosine has not been observed. Incomplete modification of the tRNA obtained in vitro must be the reason why such tRNA is not aminoacylated. Transcription of the promoter-containing synthetic gene is controlled by a promoter 51 b. p. long. The produced transcript begins with the 5'-terminal pppQ, and the entire 5'-terminal sequence corresponds to what was expected.The tyrosine suppressor tRNA gene synthesized by the chemoenzymatic procedure was built into two vectors: plasmid (ColE1) and a specially constructed vector Charon 3A based on bacteriophage
l. Both vectors had amber mutations. The incorporation into the vector followed its digestion with restriction endonuclease EcoRI.After the transformation of E. coli with recombinant DNAs carrying the synthetic suppressor tRNA gene, amber mutations were suppressed, both bacterial and in the case of bacteriophage
l; this indicates that the processing of pre-tRNA in vivo is complete and yields mature tyrosine suppressor tRNA.After cloning, the synthetic gene was separated from the vector by treatment with restriction endonuclease EcoRI and underwent analysis, including determination of its capacity to suppress bacterial mutations after incorporation into the chromosome of E. coli as part of bacteriophage
l.The transcription of the cloned gene in vitro yielded a product which was converted, after treatment with a crude E. coli extract, into a precursor of suppressor tRNATyr. The structure of the latter was confirmed by in vitro digestion by RNAse P to a 41-unit 5'-terminal precursor fragment. It turned out that the cloned gene had the structure of the natural one, as was intended by the synthesis, its expression was controlled by the promoter, and it was functionally active.
This experiment demonstrated already in the late seventies that high-molecular weight polynucleotides identical to the corresponding portions of natural DNAs can be synthesized.
In 1975, Köster and coworkers synthesized the first protein gene in a similar fashion. What they actually produced was a "minigene" or, to be precise, the gene of the peptide hormone angiotensin II. Unlike Khorana, who had synthesized his gene proceeding from the known primary structure of tRNA
and established sequence of the promoter and terminal segments, Köster was guided by the amino acid sequence of angiotensin II. The genetic code was used to establish the nucleotide sequence of the structural gene. The "minigene" also included a translation initiating segment (ATG) and two stop codons.

As was intended by the experimenters, the presence of a stop codon before the structural gene had to ensure the onset of translation.
In 1977, Itakura and coworkers synthesized a second minigene - that of somatostatin (see scheme below):
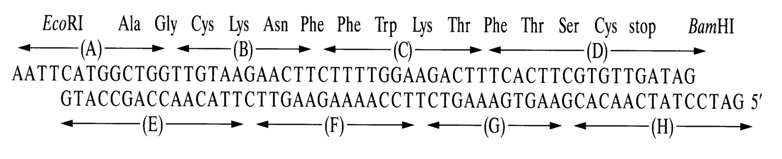
Shown on top of the scheme is the primary structure of somatostatin, while the bottom structure corresponds to the structural gene of the peptide hormone. The parenthesized letters from A to H stand for each of the 9 oligonucleotide segments synthesized by the triester method. The codons corresponding to each amino acid have been selected proceeding from the primary structure of the peptide hormone. The selection was not random but based on the frequency at which the codons occurred in the genome of phage M2. The double-stranded polynucleotide contained a methionine codon at the 5' end of the coding sequence (to ensure directed cleavage of the polypeptide at this point after translation) and had sticky ends corresponding to restrictases EcoRI and BamHI. The somatostatin gene was inserted, within a specially constructed plasmid vector, into the
b-galactose operon of the b-galactosidase gene. Thus, the regulatory apparatus of the b-galactosidase gene was used for regulating the synthetic gene. Amplification of the selected plasmid carrying the peptide gene in E. coli brought about generation of a chimeric protein (b-galactosidase bound via methionine to somatostatin) from which the peptide was separated using cyanogen bromide. The somatostatin produced in this manner was identical to its natural counterpart in terms of antigenic properties.Itakura's work can be regarded as seminal. To begin with, for the first time a bacterial cell was made to produce animal protein. Secondly, the expression of the synthesized somatostatin gene testified to correct planning of the synthetic procedure and the experiment as a whole. Thus, Itakura's efforts have opened up new possibilities in the synthesis of physiologically active peptide genes and making of peptide and protein producers.
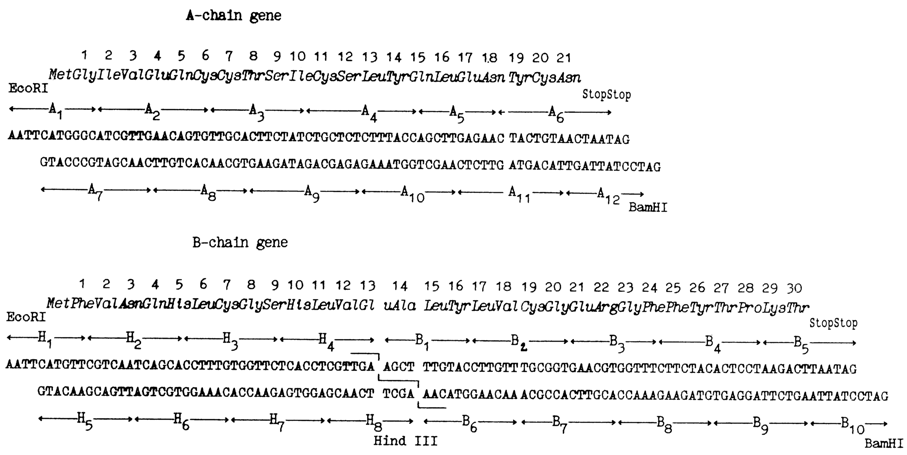
A logical extension of the work done by Itakura and coworkers was synthesis of the human insulin gene, accomplished by the same group in 1978. As is known, the insulin molecule consists of two polypeptide chains A and B. In separate syntheses, genes of both chains were produced, their primary structure being predetermined, just as in the case of somatostatin, from the amino acid sequence of the corresponding chains of insulin (see scheme 551~1.gif). It can be seen that in addition to the structural portion both genes comprise codons of methionine, stop codons, and sticky ends corresponding to restriction endonucleases EcoRI and BamHI. In the middle of the chain B gene there is a site recognized by restriction endonuclease HindIII (AAGCTT).
The synthesis of both genes had been completed within a record period of time: it took a mere six months for four workers to synthesize 77 and 104 b.p. duplexes. The synthetic work was distinguished by use of trinucleotides as "building blocks". A library of 45 trinucleotides was created within three months.
Both genes were inserted within different plasmid vectors into the
b-galactosidase segment and, after amplification within E. coli, chains A and B of insulin were produced separately in purified form to synthesize the protein itself.The next achievement by the same group was the synthesis in 1979 of an even more complex gene of the human growth hormone somatotropin. This protein consists of 191 amino acids, and in spite of the spectacular advances in the triester block method synthesis of its gene would take much more time than that of the insulin gene. Therefore, the synthesis was performed only partially, the major portion of the gene being produced through reverse transcription of mRNA from somatotropin extracted from the pituitary. However, such a cDNA could not be used in its entirety to produce the hormone because it had a leader sequence, and the corresponding polypeptide is detached from the protein only in human cells (naturally, a bacterial cell lacks the enzymes necessary for processing of the human gene). This is why the cDNA was treated with restriction endonuclease, and part of the required sequence was removed (69 b.p.) then restored using a synthesized polynucleotide having an initiation codon and a methionine one. The rest of the experiment was conducted as described above with the difference that the gene was inserted directly into the promoter. The hormone yield turned out to be quite high - 3 mg per liter of the culture fluid, although its presence was proved only by radioimmunoassay.
As automation proliferated more widely in the eighties, the number of synthesized genes increased from one year to another. 1000 b.p. genes became routine. Whatever complications were involved had to do with their composition and assembly techniques. Here is how the bovine rhodopsin gene was assembled by Khorana and coworkers in 1986.
The gene is divided into three fragments. Fragment EX contains nucleotides 5 to 338, fragment XP, 335-702, and fragment PB, 699-1052. The synthesized oligonucleotides of each fragment are numbered (the numbers are indicated above and below duplexes). The vertical arrows mark nucleotides that were replaced in comparison with the natural gene. The unique restriction sites in the synthetic gene are in solid squares. The restriction sites unique in the fragments but not in the gene itself are in dashed squares. Finally, the restriction sites present in the natural gene and removed from the synthetic one are in cross-hatched squares. Site DdeI in fragment PB has Latin letter designations because there is yet another site DdeI which overlaps with the unique site BamHI. Therefore, the second site DdeI is not recognized if the fragment containing it is treated with the enzyme BamHI.
Table 11-3A and table 11-3B illustrates genes of various length, obtained before 1988 from synthetic oligonucleotides.
The table lists not only the methods used for chemical synthesis of oligonucleotides but also some details of how the gene sequences were divided into fragments as well as procedures for their assembly. Besides, some details of cloning are also included, such as types of vectors used, structure of promoters and, wherever relevant, fusion proteins.
References to sources are given for those interested in more detailed information about synthesis of a particular gene. Seliger's laboratory (Ulm, Germany) possesses information on all genes synthesized before 1988. It is summarized in his paper in AP, 16, 7763-7769 (1988).
Conclusion
The importance of organic synthesis of DNA fragments has been realized for quite some time. As early as the first brilliant work by Khorana, in which decoding was done by direct experiments based on synthetic oligonucleotides, it became absolutely clear that directed synthesis of DNA fragments (practically the only way to obtain nucleotides with a strictly predetermined structure) would be a mainstay of molecular biology for years to come.
Several reviews offer detailed discussion of work in which relatively short synthetic oligonucleotides with a predetermined primary structure are employed to solve certain problems in molecular biology and genetic engineering.
In the eighties, a large number of rather extended protein genes, regulatory portions of genes, and similar genetic structures had been synthesized. Evidently, further advances in the synthesis of genes and their fragments in combination with proven techniques of genetic engineering will make it possible not only to obtain producers of biologically active peptides and proteins as well as easily reproduce the newly created genetic information, but also make broad use of the latter to study the functioning mechanisms of genes. It is becoming feasible to plan experiments aimed at elucidating such fundamental aspects of molecular biology as functioning of genes, recognition of different gene segments by respective regulatory proteins, polymerases, processing enzymes, hydrolases, and the like. Using natural DNA fragments for this purpose is most unlikely in the foreseeable future. Even if recourse is had to this approach, it will not provide experimenters with a set of genetic structures differing in the chemical makeup of individual fragments. Preplanned creation of genetic structures is possible only through organic synthesis, controlled replacement of natural monomers or their fragments by various analogues, inversion, insertion, fission, and other procedures.
All this stimulates organic chemists to constantly improve chemical synthesis methods. Before the eighties, organic synthesis of DNA fragments could be accomplished only in very few laboratories. With the advent of automation, organic synthesis is becoming routine - it takes much less time and methods are more easily reproducible.
Currently, genes comprising up to 2000 base pairs are being synthesized. Gene synthesis along with site-directed mutagenesis have become essential prerequisites for progress in genetic engineering as means for producing innumerable genes with single and directed substitutions and, consequently, sets of proteins with different amino acids in particular positions. These techniques will also make it possible to synthesize proteins yet to be isolated, perhaps even nonexistent, in nature. New vistas are being opened up in the development of proteinaceous catalysts for new reactions as well as all kinds of proteins to meet daily needs of man.