![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
9.7 Chemical Probing of Nucleic Acid Structure
We shall now discuss the use of reactions aimed at chemical modification of heterocyclic bases and the sugar-phosphate backbone of nucleic acids for studying their macromolecular structure. As was already mentioned in the beginning of this chapter, the reactivity of a particular nucleotide in DNA or RNA is to a certain degree dependent on the secondary or tertiary structure element of the nucleic acid accommodating it. Consequently, by studying the degree of modification of particular functional groups of bases as well as the carbohydrate moieties and phosphate group of each nucleotide in a nucleic acid molecule one can obtain valuable information about its three-dimensional structure. This approach has become known as chemical probing. In combination with other physical methods, it allows the macromolecular structure of DNA and RNA to be described with high resolution. Moreover, by comparing the reactivity of nucleotides in DNA or RNA in a free state and in a complex with specific ligands, one can identify, also with a high resolution, nucleic acid sites responsible for specific binding and recognition of a particular ligand.
Chemical probing is especially valuable in cases where X-ray structural analysis or NMR spectroscopy are not applicable, for example, when analysing large RNAs or multicomponent complexes of nucleic acids with proteins. Furthermore, in contrast to X-ray structural analysis, chemical probing makes it possible to study the behavior of nucleic acids in solution as well as the dynamics of their interaction with proteins over the course of time. Another important consideration is the fact that the sample amount for this technique can be hundreds or even thousand of times smaller than for many physical measurements.
9.7.1 Location of Modified Nucleotides
The improvement of methods for locating modified nucleotides in DNA and RNA has gone hand in hand with advances in nucleic acid sequencing because the objectives are similar or identical in both cases.
Whereas sequencing of nucleic acids had been based on identification of the primary structure of each fragment, in order to identify a modified base one also had to find the host oligonucleotide, then isolate the modified base by further cleavage and fractionation. Therefore, the search for a particular modified nucleotide had always been an extremely difficult task. This inevitably led to disturbance of its macromolecular structure and, ultimately, to wrong conclusions. A case in point is attempts to determine the tertiary structure of tRNA in the years preceding elucidation of its crystal structure. The chemical modification data had formed the basis of many models of the tRNA tertiary structure proposed at that time, and every model turned out to be incorrect.
The situation changed drastically with the development of rapid methods of gel sequencing of DNA and RNA (see Chapter 6). Today, the following two strategies of modification and location of modified nucleotides are used (see Fig. 9-1).
In the first case, just as during nucleic acid sequencing by the Maxam-Gilbert method, subjected to modification is a homogeneous nucleic acid, or its fragment, labeled at one end (usually with radioactive phosphate). The reaction is conducted in such a manner that not more than one nucleotide, on the average, becomes modified in each nucleic acid molecule, which is achieved by lowering both the concentration of the reagent and the reaction temperature. The direction of the reaction is thus determined only by the starting three-dimensional structure of the nucleic acid. Then the polynucleotide chain is cleaved at the modified base (if the cleavage does not take place during the modification itself, for example, when "chemical nucleases" are used), and the length of the resulting fragment, measured during gel electrophoresis, is used to locate the base to within a single unit.
As can be seen, the first strategy is restricted to use of reagents modifying DNA or RNA so that it can be specifically cleaved and to homogeneous polynucleotides with a length not exceeding 300 to 400 nucleotides. These restrictions are removed in the second strategy of locating modified nucleotides, which has turned out to be especially useful in studying large RNAs.
In this case, an oligodeoxyribonucleotide complementary to a nucleic acid segment located not far from and downstream of the target sequence is synthesized. After the modification reaction is over (it is also conducted so as to modify not more than one base), the oligonucleotide is used as a primer for reverse transcriptase. If the chemical modification of nucleotides is carried out in a way that inhibits Watson-Crick hydrogen bonding, the enzyme will not be able to read modified bases and will stop at a modification site (Fig. 9-1). Of course, it will also stop if the modification leads to rupture of the polynucleotide chain. Then, the length of the synthesized cDNA, determined by means of dideoxy sequencing ladders (see Chapter 6), is used to locate the modified nucleotide. And if the modification involves a single-stranded DNA, one of the DNA polymerases is used instead of reverse transcriptase.
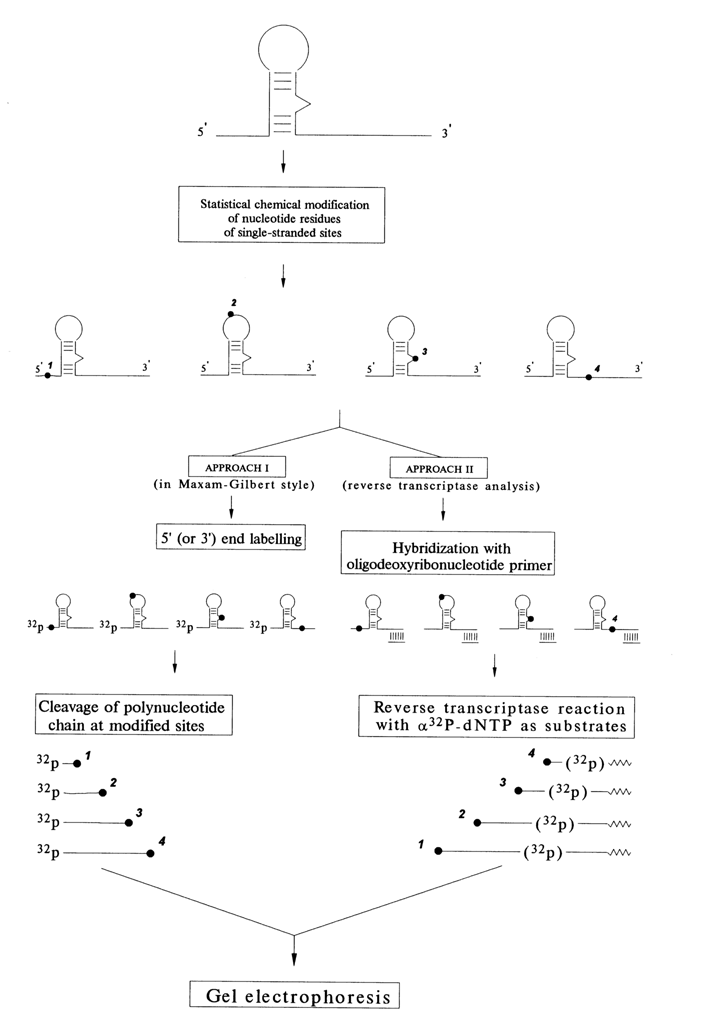
Fig. 9-1. Two strategies of localization of modified nucleotide residues in a nucleic acid molecule.
Optimally, both strategies enable one to monitor every base over the nucleic acid segment under investigation. Some problems may arise in interpretation of the results. In particular, it is not always clear why a particular nucleotide is not reactive or, to be more precise, whether it is involved in secondary or tertiary interactions. Investigating RNAs under partially denaturing conditions, when the tertiary structure is disturbed to some degree whereas the secondary is yet intact, often helps in interpreting the results of chemical probing.
The same strategies are used in studying nuclein-protein complexes. To this end, the chemical modification of DNA or RNA in a free state is in most cases compared with that in a complex with a protein. In this way it becomes possible to detect the trace left by the protein on the nucleic acid molecule, which is why this technique has become known as footprinting.
9.7.2 Systematic Chemical Probing of RNA Secondary and Tertiary Structure
9.7.2.1 Transfer RNA
Chemical probing in combination with gel sequencing was used for the first time by Peattie and Gilbert in 1980 to monitor the tRNAphe conformation in solution. They employed two reagents - dimethylsulfate (DMS) to probe position N7 of guanine and N3 of cytosine as well as diethylpyrocarbonate (DEPC) to probe position N7 of adenine (for mechanisms of these reactions, see 9.2.2.2). Since the use of these reagents allowed the investigators to subsequently cleave phosphodiester bonds at modification sites, tRNA was labeled with 32P at one end and the first strategy shown in Fig. 9-1 was used to locate a modified base (see Fig. 9-2). The result was information about 51 out of 76 bases in tRNA. It had been established that most bases behave as could be expected from the crystallographic three-dimensional structure of tRNA. However, at least three bases (A73, C72 and G19) did not fit the pattern. and eight guanines were assumed to have been involved in hitherto unknown tertiary interactions.
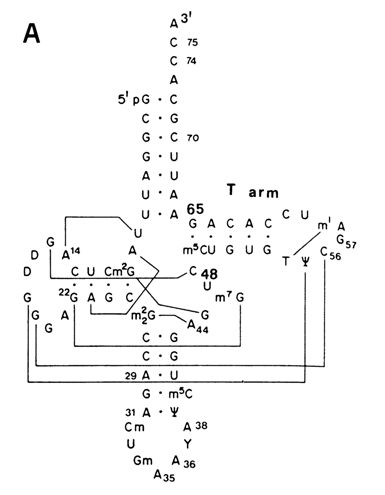
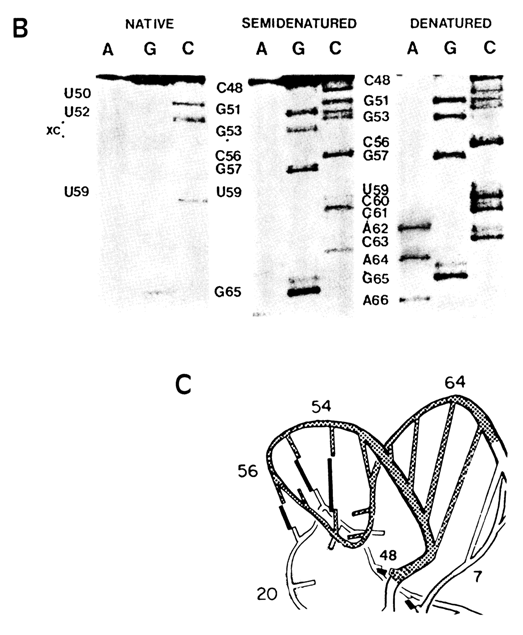
Fig. 9-2. Secondary and tertiary structure of the T arm part of
tRNAPhe according to the results of modification with DMS and DEPC.
A. The cloverleaf structure of yeast tRNAPhe; lines connect the bases involved
in tertiary contacts.
B. The portions of gel autoradiograms illustrating chemical probing of the native,
semidenaturated and denaturated forms of the T arm.
C. Tertiary structure of the T arm part of yeast tRNAPhe (shaded area);
tertiary base-base hydrogen bonds are marked by black rods (adapted from D. A. Peatie and
W Gilbert, Proc. Nat. Acad. Sci. USA, 77, 4679-4682 (1980)).
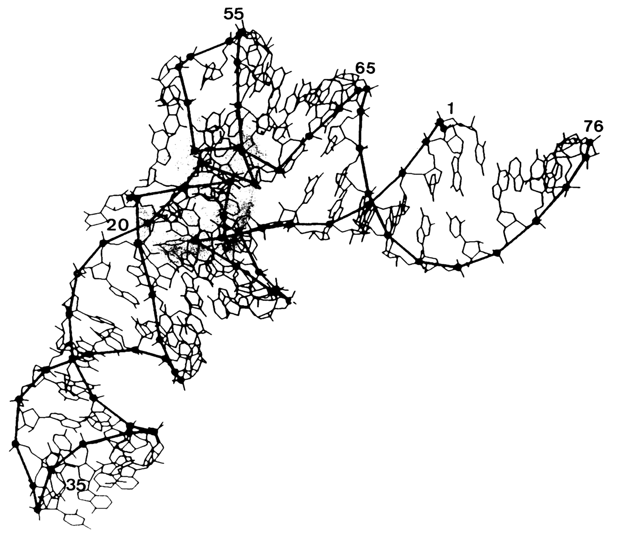
Fig. 9-3. Three-dimensional structure model of initiator E. coli tRNAMet based on the results of the detailed chemical probing; the shaded area is the region where phosphate groups are protected from the attack with ethylnitrosourea (adapted from H. Wakao et al.,J. Biol.Chem., 264, 20363-20371 (1989)).
Later, many tRNA were subjected to chemical probing. Figure 9-3 illustrates the results of monitoring of E. coli initiator tRNA. In this particular case, added to the above-mentioned DMS and DEPC was 1-cyclohexyl-3-(2-morpholinoethyl)-carbodiimide metho-para-toluene sulfonate (CMCT) which modifies Watson-Crick positions N1 in guanine and N3 in uridine. Moreover, adenines were monitored by DMS at N1. A radically new approach here is monitoring of phosphates by ethylnitrosourea (ENU). Modifications at phosphate groups as well as at C(N3), G(N7) and A(N7) were identified after tRNA cleavage. Modifications at A(N1), G(N1) and U(N3) could only be identified by primer extension. Combination of all these reactions has given a good insight into the tertiary structure of initiator tRNA, which has turned out to be instrumental in studying the interaction between this tRNA and proteins involved in protein biosynthesis.
Such a detailed picture provided by chemical monitoring of tRNA can only be enhanced by the results of tRNAPhe cleavage by Fe(II)-EDTA (see also 9.5.2). Fe2+, ions in a complex with EDTA in the presence of O2 and a reducing source (e.g., dithiothreitol) generate hydroxyl radicals attacking ribose in RNA and deoxyribose in DNA. The attack occurs near the site of binding of the redox-active metal complex with the nucleic acid. As can be inferred from Figure 9-4, several short tRNA segments are protected against "chemical nuclease". Just as expected. the tertiary interactions observed in this experiments are stabilized by magnesium ions.
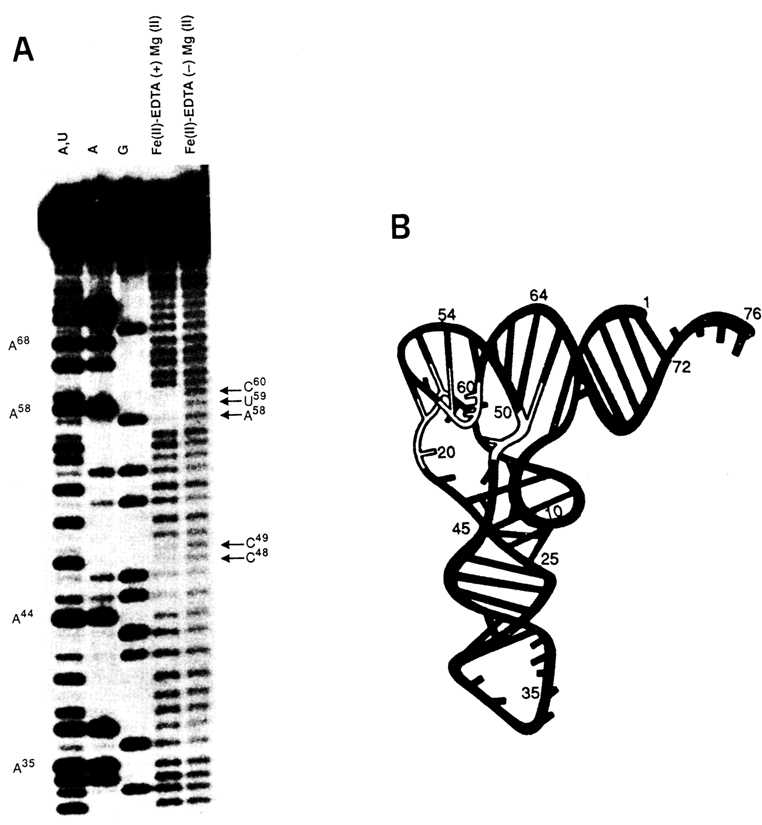
Fig. 9-4. Cleavage of tRNAPhe by Fe(II)-EDTA.
A. Autoradiogram of the polyacrylamide gel; A+U, A and G are sequencing lines; oxidative
degradation of the tRNA chain is clearly seen in the lane corresponding to the native form
of tRNAPhe (marked with arrows).
B. Three~dimensional structure of tRNAPhe ; white backbone indicates protection
from Fe(II)-EDTA. (adapted from J. A. Latham and T. R. Chech, Science, 245, 276-282
(1989)).
9.7.2.2 Ribosomal RNA
We have already mentioned that the secondary structure models for all types of RRNA are based primarily on comparative (phylogenetic) analysis of the primary structures of rRNAs in many organisms (see 8.2.3). These models were experimentally tested and refined by chemical probing.
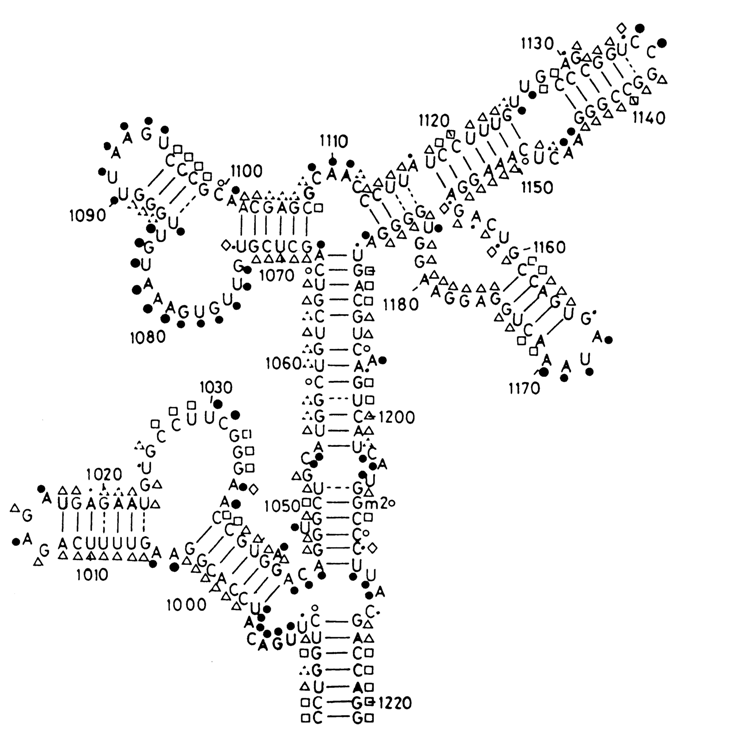
Fig. 9-5. Scheme illustrating the exhaustive chemical modification of the segment (nucleotides 984-1221) of the 16S RNA from E. coli ribosomes by DMS and CMCT. Reactive under native conditions: (
·) strong hit, (·) moderate hit, (·) marginal hit; unreactive under native conditions but reactive under semi-denaturing conditions: (D) moderate hit, (\) marginal hit; unreactive under both native and semi-denaturing conditions: (‚); (?) denotes increased reactivity under semi-denaturing conditions, as compared to native conditions. (°) denotes not determined positions (reproduced with permission from C. Ehresmann et al., Nucl. Acids Res., 15, 9109-9128 (1987)).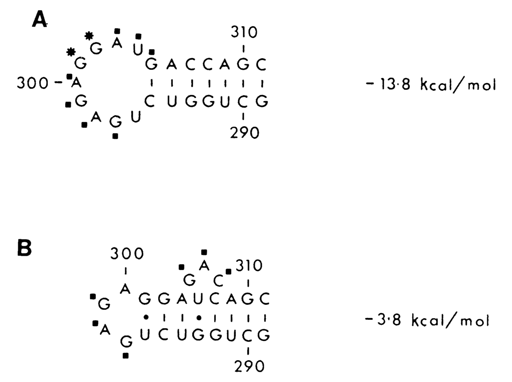
Fig. 9.6. Two secondary structures of the 290 to 310 region of the 16S RNA from E.
coli ribosomes supported by chemical probing (* - strong hits, ·
- moderate hits)
A. The segment in the naked 16S RNA.
B. The same segment in the 30S subunit
(adapted from D. Moazed, S. Stern and H. Noller, J. Mol. Biol., 187, 399-416
(1986)).
Chemical monitoring of the RRNA structure was most widely used in Noller's and Ebel's laboratories. In addition to the reagents mentioned in the previous section use was also made of kethoxal (KE) which modifies guanine bases at N1 and N2. The modified bases were identified by the primer extension approach. Used for this purpose was a series of oligodeoxyribonucleotides that prime reverse transcription at 150-200 nucleotide intervals along the RNA chain. As a result, it became possible to monitor the overwhelming majority of bases in rRNA and to demonstrate convincingly that the theoretical models quite adequately describe the macromolecular structure of RRNA both in a free state and as part of ribosomal subunits (see Fig. 9-5).
It should be mentioned that reverse transcriptase makes quite a few spontaneous stops on unmodified rRNA. Although they make it more difficult to interpret the results of chemical probing, every such stop carries additional information about the rRNA structure, insofar as it is indicative of incorporation of the nucleotide in question into a stable element of the secondary or tertiary structure of rRNA.
Another example, shown in Figure 9-6, illustrates how chemical probing can be used to corroborate a local conformational change occurring in the short hairpin loop of E. coli 16S rRNA forming part of the 30S ribosomal subunit. Interestingly, this rRNA secondary structure element acquires a conformation which is energetically less advantageous.
The method just described has found broad application in monitoring the structure of large RNAs of other types, including viral and messenger RNAs.
9.7.2.3 Searching for Unusual RNA Structures
Quite often, chemical probing reveals unusual features in RNA structure, such as non-Watson-Crick base pairing.
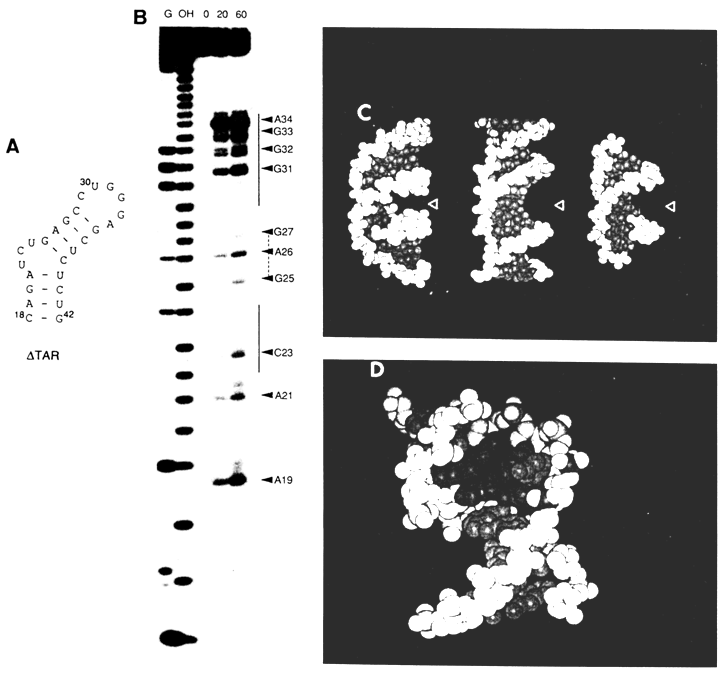
Fig. 9-7. A. Secondary structure of
DTAR.Let us now consider a case where the chemical modification method was instrumental in bringing to light an unusual conformation in a short double helix forming part of TAR - transactivation response RNA sequence of the human immunodeficiency virus type 1 (HIV-1). TAR is located at the 5' end of the untranslated leader region of all viral messenger RNAs. It comprises nearly 60 nucleotides and has a stable stem-loop structure. The TAR segment consisting of 27 nucleotides and marked
DTAR (Fig. 9-7A) forms a specific complex with HIV-1 Tat protein, and this binding is essential for virus replication.The conformation of DTAR was studied by subjecting it to chemical modification by DEPC. It has already been mentioned that DEPC is used for monitoring adenines at N7. DEPC also reacts, albeit less effectively, with N7 in guanines. If adenine or guanine take part in Watson-Crick base pairing, their N7 are unreactive because they are buried deep in the narrow major groove of the RNA helix. Besides, DEPC reacts to some extent with pyrimidine bases as well, although the mechanism of this reaction is yet to be elucidated.
It can be seen from Figure 9-7A that DTAR contains a bulge consisting of three nucleotides. In experiments with modification by DEPC two mutant structures were used in addition to DTAR. One of them had no trinucleotide bulge at all, whereas in the other bases 23 and 24 were removed from the bulge. The chemical modification results indicate (Fig. 9-7B) that the presence of a bulge radically alters the conformation of the adjacent double-helix segments. If in the bulge-free mutant
DTAR base-paired purines were unaffected by DEPC treatment, just as expected, in the wild type variant of DTAR several purine bases were modified at the atoms that were supposed to occupy the major groove of the double helix. To explain this inconsistency it was speculated that the A form of the RNA double helix in DTAR acquires a conformation which looks more like the B form (Fig. 9-7C,D). In this case, the major groove widens and positions N7 in purines become accessible to DEPC.9.7.3 Studying Conformational Changes in DNA
As already mentioned in Chapter 8, the double helix of DNA is in B form under normal conditions. However, if DNA in a superhelical state contains certain specific nucleotide sequences, its macromolecules may give rise to unusual structures (Z form, cruciform, H, form [see 8.1.7]).
The chemical modification method has turned out to be a powerful tool for identifying such forms.
It should be remembered that in the early experiments in which cruciforms were revealed in a palindromic DNA use was made of nuclease S1 which cleaves single-stranded DNA segments inevitably produced during formation of cruciforms (Fig. 8-13). It might be assumed, however, that the protein interacting with the DNA somehow stabilizes the cruciform. Therefore, taken as direct proof of emergence of such a form could be only modification of the corresponding DNA loop segments by reagents specific toward single-stranded polynucleotides.
For instance, a cruciform was found in a plasmid DNA, whose single-stranded segment was expected to contain cytosines, as a result of their conversion into uracils by sodium bisulfite treatment (i.e., as a result of oxidative deamination). This plasmid was used to transform E. coli cells deficient in uracil-N-glycosidase. After replication of the plasmid, U . G pairs were substituted by T . A ones. These mutations were detected further by sequencing the corresponding DNA segments. The same experiments also demonstrated that the cytosines at the junction between the four helices of the cruciform were not modified by bisulfite, the implication being that the DNA helices are joined together without unwinding.
A simpler way to find single-stranded loops in a DNA cruciform is through carboxyethylation of positions N7 of their purine bases with DEPC, followed by cleavage of the chain at the modification sites. Modification of purine bases by DEPC does not reveal any single-stranded segments at the junction between the four helices either. Moreover, it has led to a discovery that the optimal size of a loop in the DNA cruciform is four to six nucleotides.
The left-handed Z form of the double helix in DNA can be detected by several procedures based on chemical modification. Firstly, the DNA segments lying at the boundary between B and Z forms are single-stranded and can be revealed with the aid of DEPC (purines), hydroxylamine (cytosines) or OsO4 (thymidines). Secondly, as has already been pointed out (see 8.1.2), pyrimidine bases in DNA segments that are in Z form are in an anti-conformation, whereas purine bases in the same segments are in a syn-conformation. As a consequence, the imidazole ring of purines is closer to the double helix surface and becomes vulnerable to DEPC attack. For example, when the d(A-C)32d(G-T)32 sequence is incorporated into a cyclic DNA and turns into a superhelical molecule with a supercoiling density necessary for transition B
®Z, A and G in this sequence start being modified. Interestingly, at the same time purines in the segments lying next to the incorporated one are modified as well. This indicates that the Z form spreads over regions lying beyond the alternating sequence. Finally, there is a reagent that can be used for directed probing of the left-handed DNA helix, namely, A-Co(DiP)33+ [tris(4,7-diphenyl-1,10-phenanthroline)cobalt (III)]; it is specifically bound with DNA in Z form and cleaves it after photoactivation. It is experiments with this particular reagent that have suggested possible presence of the Z form in functionally important DNA segments.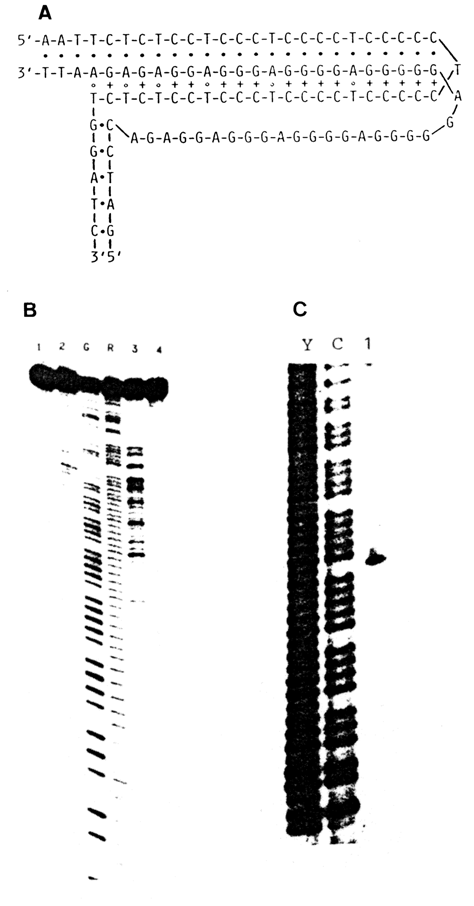
Fig. 9-8. Chemical probing of H form of DNA.
A. H form of the DNA segment incorported into a superhelical plasmid.
B. Autoradiogram showing the results of modification of the DNA segment with DEPC (line
3).
C. Autoradiogram showing the results of modification of the DNA segment with OsO4.
Modification of the T residue in the loop is clearly seen (line 1) (courtesy of O. N.
Voloshin).
Since transformation from the B into the H form of DNA is accompanied by formation of a triple helix so that one of the polypurine strands is in a single-stranded state (Fig. 8-15), it is absolutely clear that this unusual form can also be detected with the aid of reagents attacking adenine and guanine only in single-stranded segments of nucleic acids. Figure 9-8 demonstrates the results of such an experiment.
9.7.4 Evaluation of Four-Stranded G4-DNA
Guanine-rich sequences, especially common in telomeric portions of chromosomes, are also found in certain recurring DNA segments, in promoters of some genes and immunoglobulin-switch regions. On several occasions we pointed out the capacity of guanine bases for forming tetrads, which is the reason for formation of four-stranded DNA regions known as G4-DNA.
As can be seen from Figure 7-27, in a G-tetrad all N7 atoms of the imidazole ring in guanines are involved in hydrogen bonding. It is therefore to be expected that they will be fully protected against modification by DMS. This simple test is used today as proof of formation of G4.
Figure 9-9 shows the results of methylation of the poly (G) insert into a circular DNA, clearly indicating that guanine-rich sequences acquire a four-stranded form.
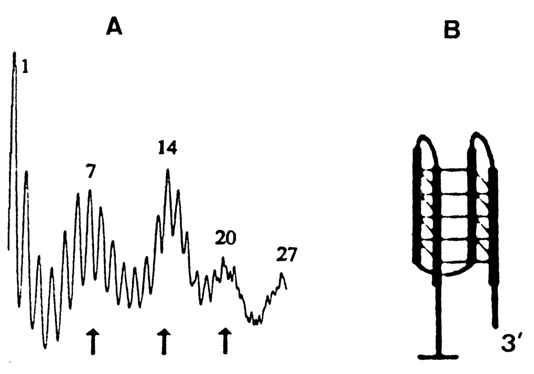
Fig. 9-9. Evaluation of G-DNA by chemical probing.
A. Distribution of guanine methylation within the (dG)27 insert into a plasmid.
B. Model of the G-DNA structure. Three loops in the structure correspond to the three
peaks (marked by arrows) in A. (adapted from I. G. Panyutineal et al., Proc. Nat. Acad.
Sci. USA, 87, 867-870 (1990)).