![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
9.5 Reactions of Nucleotide Phosphate Groups
The reactions involving internucleotide phosphate groups may either be accompanied by degradation of the polynucleotide chain (as a result of cleavage of internucleotide linkages) or leave the latter intact.
9.5.1 Cleavage of Internucleotide Linkages
Obviously, the stability of a polymeric nucleic acid molecule is determined by the reactivity of the internucleotide node in aqueous solutions. It is most important for an experimenter working with nucleic acids to know the limits of stability of internucleotide linkages in both DNA and RNA. To a certain degree, this has to do with the fact that the result of biological research often hinge on the integrity of the native primary structure of the biologically active nucleic acid molecule: cleavage of just one of a thousand internucleotide linkages may lead to a complete loss of activity. Thus, reactions leading to cleavage of phosphodiester (internucleotide) bonds occupy a special position among those involving nucleic acids.
Reactions in which the polynucleotide chain undergoes degradation may be based on two different mechanisms. Reactions of the first type, with cleavage of the P-O bond, are essentially nucleophilic substitution at the internucleotide phosphorus atom which, in this case, exhibits electrophilic properties.
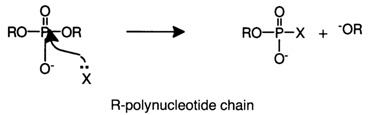
The nucleophilic attack of the phosphorus atom may be inter- or intramolecular.
Reactions of the second type, with cleavage of the C-O bond, occur when a carbonyl group emerges in the dinucleoside phosphate fragment of the polynucleotide at the
b-position with respect to the phosphodiester group. They take the form of b-elimination catalysed by bases: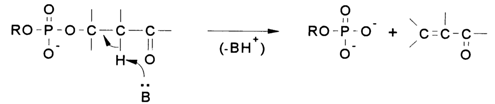
Just as with cleavage of the P-O bond, the motive force behind the
b-elimination is the electrophilic behavior of the phosphorus, the capacity for cleaving the C-O bond being in this case determined by the stability of the departing anion. Consequently, this type of cleavage of internucleotide linkages is indirect, dependent on some kind of primary transformations giving rise to a carbonyl at the b-position with respect to the internucleotide phosphodiester group. This is the mechanism governing acid hydrolysis of DNAs, hydrolysis of depyrimidinated RNAs by primary amines, as well as detachment of the 3'-terminal nucleosides in RNAs after periodate oxidation.Reactions of Nucleophilic Substitution at the Intemucleotide Phosphorus Atom (cleavage of the P-O bond). The stability of internucleotide linkages in RNAs and DNAs has already been discussed at length. It was pointed out that the sharp difference between the two in this respect stems from the presence or absence of a hydroxyl at the C2' atom next to the internucleotide node.
Internucleotide linkages in DNAs are extremely stable in both acid and alkaline media. In an acid medium, their hydrolysis takes place at pH < 3. As regards alkaline media, the hydroxyl ion attack at the phosphorus atom is hindered, just as in the case of simple dinucleoside phosphates, by the negative charge at the phosphate group (pKa1 of the phosphate group does not exceed unity at pH > 3, and the internucleotide phosphate group is ionized almost completely). Internucleotide linkages in RNAs are much less stable. If you remember, this is due to the presence of a hydroxyl at position 2' in ribose, which acts as an internal nucleophile in this case. The cis-configuration of the glycol group in the furanose ring of ribose is the reason for the ease of nucleophilic substitution even at pH values that are not too alkaline. Already at pH 10, internucleotide linkages in RNAs start breaking, which is indicative of the fact that this process has to do with dissociation of the 2'-hydroxyl group (pK~12).

The rate of this process is strongly influenced by the nature of the nucleotides forming the dinucleoside phosphate fragment under investigation. This influence seems to be due to stacking interactions and the presence of charged groups in the heterocycles. For instance, studies into the rate of alkaline hydrolysis of internucleotide linkages in dinucleoside phosphates have shown that they are more stable in purine derivatives and less stable in pyrimidine ones where stacking is not pronounced. Stacking interactions are likely to result in such a conformation of the internucleotide node when the phosphorus atom is remote from the OH group at C2' and the rotation about the C3'-O bond is slowed down. This inhibits the formation of the pentacoordinate phosphorus in the transient state, emerging during intramolecular nucleophilic attack of the internucleotide phosphate group by the 2'-hydroxyl. The rate of hydrolysis of interribonucleotide linkages suggests that stacking reduces the rate of their cleavage by one order of magnitude.
Table 9-3. Partial Hydrolysis of RNA in an Alkaline Medium Under Different Conditions.
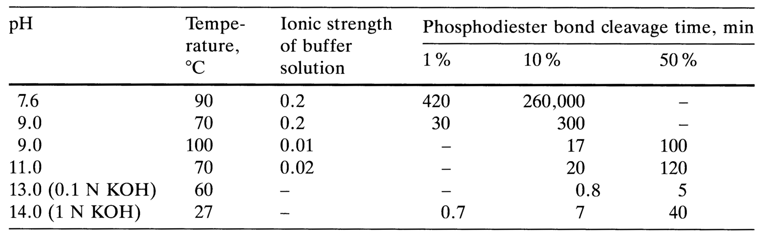
The difference in the rates of interribonucleotide linkages is maximum at pH 11, when the ionization of uracils and guanines upsets the course of stacking interactions. Under such conditions, polyguanyl fragments are hydrolysed faster than polyadenyl ones and polyuridyl fragments are hydrolysed faster than polycytidyl ones.
The alkaline hydrolysis of RNA to mononucleotides is usually carried out using a 0.3 N KOH solution at 370 C for 20 h. Under milder alkaline conditions, RNA yields oligonucleotides. Table 9-3 summarizes the effect of such factors as pH of the medium, temperature and ionic strength on the rate of hydrolysis of internucleotide linkages in RNA.
Ions of many heavy metals exert catalytic action on the hydrolysis of internucleotide linkages in RNA, catalysis being the most effective at neutral and weakly alkaline pH - that is, under conditions conducive to formation of metal salts with phosphate groups. Such formation of salts (complexes) seems to promote the nucleophilic attack of the phosphorus atom by the hydroxyl at C2, which is possibly involved in the complex formation as well, whereby its nucleophilic activity is enhanced.
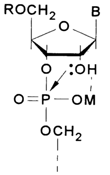
The participation of the neighboring 2'-hydroxyl in this process is inferred from the fact that DNA is not hydrolysed under similar conditions; RNA methylated at 2'-OH groups is not hydrolysed either.
Comparison of the catalytic activity of ions of different metals suggests that compounds of lead and lanthanum are the most active catalysts. Ions of bivalent zinc and cadmium are less active. The rate of RNA hydrolysis in the presence of ions of heavy metals is also materially affected by the nucleotide composition of RNA. Purine polynucleotides are hydrolysed more slowly than pyrimidine ones. Hydrolysis of RNAs with a more stable secondary structure proceeds at al slower rate. The effect of ions of heavy metals on the strength of internucleotide linkages in RNAs should be taken into account in experiments with biologically active molecules even in cases where the experimental conditions are rather mild (neutral pH values, room temperature). For example, ions of bivalent lead, nickel and cobalt (10-3-10-4 M, pH 7, 200 C) catalyze the polymerization of RNA from tobacco mosaic virus, yeast RNA, and other polyribonucleotides.
Intermolecular attack of the internucleotide phosphorus by a nucleophile may also lead to degradation of the polymer. For instance, treatment of RNA with a 1 N solution of sodium methylate in a mixture of dimethylformamide with methanol yields ribonucleoside 3'(2')-methyl phosphates.

The mechanism of such hydrolysis may include not only intermolecular attack of the phosphorus by the methylate ion but also that of the nucleoside 2',3' cyclic phosphate forming in advance.
In an acid medium (pH
£ 3), RNA is also hydrolysed to a mixture of ribonucleoside 3'(2)-phosphates, the mechanism of such hydrolysis being similar to that of alkaline rupture of RNA. A distinctive feature of acid hydrolysis of RNA is the simultaneous isomerization of the internucleotide (3'-5' « 2'-5) linkages. The factors responsible for such isomerization have already been discussed in the context of alkyl phosphate migration in ribonucleoside 3'(2') alkyl phosphates. The rates of acid hydrolysis and isomerization of internucleotide linkages in RNA are strongly influenced by the nucleotide composition, this influence being similar to that in the case of alkaline hydrolysis already described.Hydrolysis of RNA in 1 N hydrochloric acid (1000 C, 1 h) is used for analytical purposes. During such treatment, cleavage of all internucleotide linkages goes hand in hand with hydrolysis of the N-glycosidic bonds in purine nucleoside 3'(2')-phosphates so that acid hydrolysis of RNA yields purines and pyrimidine ribonucleoside phosphates. Quantitative analysis of these compounds forms the basis of the method for determining the nucleotide composition of RNA.
Table 9-4. Partial Hydrolysis of RNA in an Acid Medium Under Different Conditions.
Acid hydrolysis of RNA may be interrupted at the step of formation of oligonucleotides which, as can be seen from Table 9-4, determine the dependence of the time it takes for internucleotide linkages in RNA to be cleaved on pH and temperature.
Isomerization of internucleotide linkages is an undesirable side process during acid hydrolysis of RNA to oligonucleotides.
b
-Elimination Reaction (cleavage of the C-O bond). It has already been pointed out that the cleavage of the C-0 bond in an internucleotide node and, consequently, rupture of the polynucleotide chain occurs when there is a carbonyl group in the carbohydrate moiety at the b-position with respect to the phosphodiester group. The reaction proceeds as P-elimination of the phosphate group under very mild conditions and almost quantitatively. The carbonyl is introduced at the first step usually in two ways: elimination of the base, which is normally used for degradation of both DNAs and RNAs, or oxidation of the 3'-terminal cis-glycol group in RNA. Let us first consider the first procedure. Hydrolysis of the N-glycosidic bond, accompanied by elimination of the base, yields an aldehyde group in the carbohydrate moiety. This may lead to b-elimination of the phosphomonoester group (polynucleotide chain with a 5'-terminal phosphate group) or, in other words, cleavage of the polynucleotide chain at the C-O bond. The reaction is catalysed by acids, alkalies and also amines.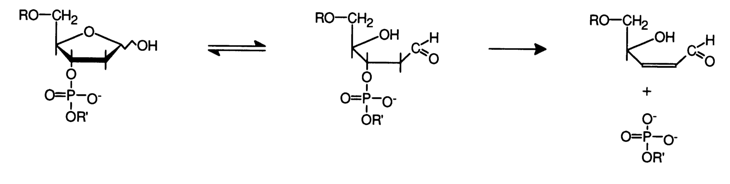
In the case of DNA, purine bases are eliminated under rather mild conditions. The resulting depurinated nucleic acids readily undergoing b-elimination under the same conditions may be isolated and subjected to exhaustive b-elimination in the presence of aromatic amines in an aqueous solution of formic acid. The above-described method for hydrolysis of DNAs to polypyrimidine units, known as the Burton method, resides in incubation of DNA with a 2 % solution of diphenylamine in 66 % formic acid at 300 C for 17 h. Under the same conditions, hydrolysis of N-glycosidic bonds formed by purine bases takes place along with b-elimination of a poly(oligo)-pyrimidine unit:
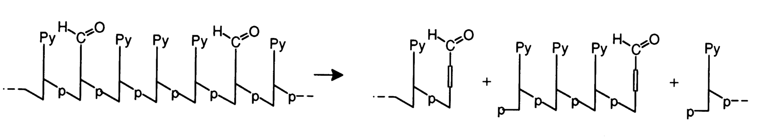
The detachment of
a,b-unsaturated aldehydes from the poly(oligo)nucleotide chain proceeds quantitatively as a result of the second elimination event. The latter involves the same mechanism as b-elimination since, in this case, the electron-acceptor effect of the aldehyde group, enhancing the proton mobility of the corresponding hydrogen atom. is to a considerable degree transmitted through the vinyl group (vinylogy).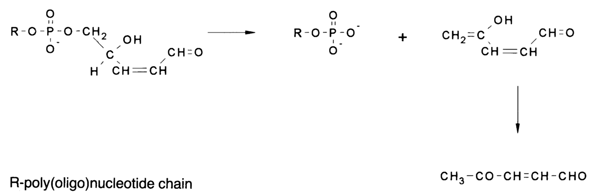
Diphenylamine, without which practically no cleavage of phosphodiester bonds is possible, seems to form a salt together with the carbonyl of the acyclic form of deoxyribose, which facilitates the
b-elimination:
Subsequently, poly(oligo)pyrimidine fragments are, as a rule, divided along the chain and then according to composition (resulting in so-called isoplates). Hydrolysates of
jX174 phage DNA have yielded pyrimidine units up to 11 nucleotides long, those of f1 phage DNA, up to 19 nucleotides, and those of fd phage DNA, up to 20 nucleotides.Alkaline treatment of depurinated nucleic acids also breaks internucleotide linkages as a result of
b-elimination of the phosphate group. However, because of the facilitated course of side reactions into which aldehydes are likely to enter in an alkaline medium, some phosphate groups remain linked to the carbohydrate moiety after b-elimination of the 3'-phosphate, for example: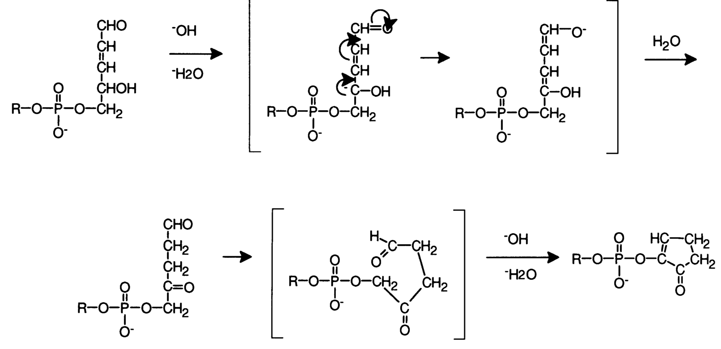
The second procedure, which permits partial elimination of pyrimidine bases from DNA, boils down to its treatment with hydrazine. The resulting depyrimidinated DNAs cannot be subjected to hydrolysis under conditions of acid catalysis because of the extremely high lability of the glycosidic bonds in purine nucleotides. This difficulty, however, has been successfully obviated by resorting to pretreatment of depyrimidinated nucleic acids with benzaldehyde (to eliminate the residual hydrazine) with subsequent degradation of the polymer under the effect of weak bases. This is how polypurine isoplates are obtained, albeit not quantitatively, under very mild conditions.
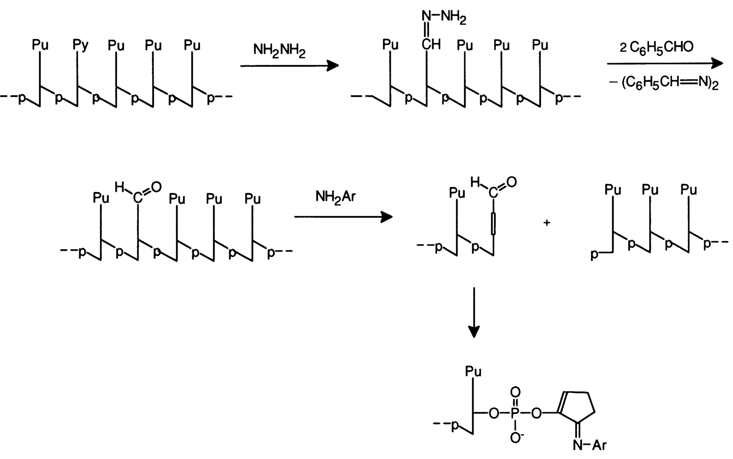
Good results were obtained when aromatic amines (aniline, para-anisidine) were used as the weak bases for
b-elimination. After separating isoplates one can derive oligopurine units which, as can be seen on the above scheme, have a blocked phosphate group at the 3' end. Similar approaches for degrading RNAs are of little practical use because internucleotide linkages in the latter are much more labile, while N-glycosidic bonds are much more stable. However, after elimination of uracils from an RNA by means of hydroxylamine at pH 10, uracil-free nucleic acids may undergo, after acid hydrolysis under mild conditions (to eliminate the hydroxyamine residue), degradation catalysed by aromatic amines (e.g., para-anisidine at pH 5). The transformations occurring in the RNA unit being hydrolysed can be represented as follows: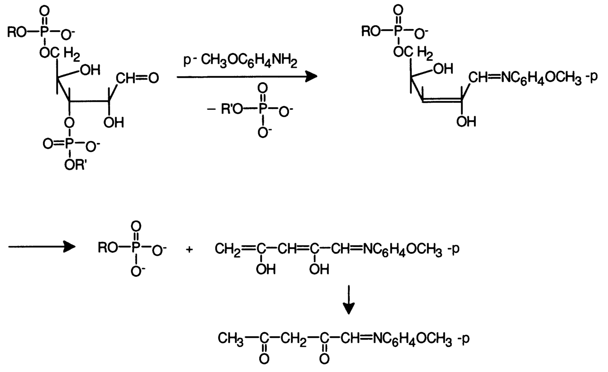
Degradation of tRNAPhe can serve as an example of controlled cleavage of the polyribonucleotide chain at a nucleotide with an eliminated base. The RNA, which is 76 base pairs long, contains base Y at position 37 (see the following scheme). This base is rather easily eliminated from tRNAPhe during hydrolysis with 0.1 N hydrochloric acid at room temperature, without any other bonds in the tRNA being affected.
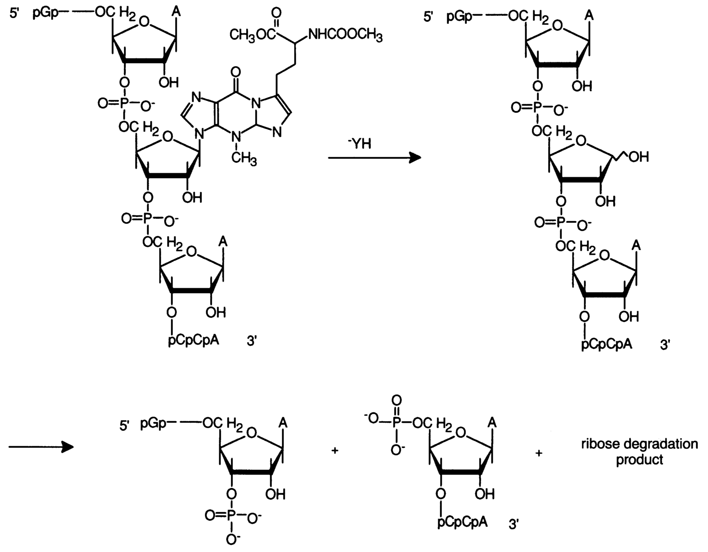
A similar degradation can also be carried out in the case of RNAs containing N7-methylated guanines and N3-methylated adenines. A common approach recommended for controlled cleavage of the polynucleotide chain at a particular nucleotide includes preliminary modification of a base in the latter with the result that the N-glycosidic bond is considerably labilized and it becomes possible to cleave both the glycosidic bond and the internucleotide linkages at this nucleotide (by the
b-elimination mechanism).The rupture of internucleotide linkages as a result of
b-elimination can be put to practical use for step-by-step degradation of RNAs from the 3' end of the polyribonucleotide chain. The first step of such a reaction is periodate oxidation of the 3'-terminal cis-glycol group. The resulting 2',3'-dialdehyde contains a 3'-aldehyde group at the b-position with respect to the 5'-phosphodiester bond. The polynucleotide unit is eliminated under the effect of various primary amines (cleavage of the C5,-O bond). As a consequence, a polyribonucleotide shortened at the 3' end is formed.
The entire cycle of transformations can be repeated. To this end, the phosphate group is enzymatically removed (with the aid of PME) from the 3'-terminal nucleotide. Acid (pH £ 3) or alkaline (pH > 11) hydrolysis of the former 3'-terminal nucleotide yields a pyrimidine or purine base that can be identified by conventional analytical methods. As has already been mentioned, this procedure is used to determine the primary structure of oligoribonucleotides. Quantitative periodate oxidation and b-elimination are achieved by conducting the reaction at pH values close to neutral. The primary amine seems to perform two functions: it participates in the formation of active intermediates and catalyses the b-elimination process. The results of studies into the effect of pH, amine structure and other factors on the course of degradation of the polynucleotide chain, based on the b-elimination mechanism, together with the data on the structure of the intermediate compounds, obtained in experiments with simpler model systems, suggest that apart from direct b-elimination this process also includes formation of some cyclic intermediates whose subsequent transformations lead to shortening of the polyribonucleotide chain by one nucleotide.
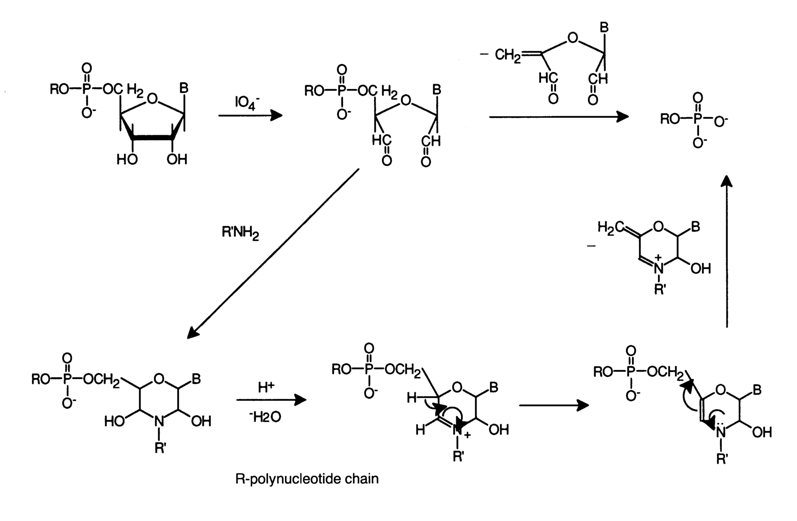
9.5.2 Cleavage of the Sugar-Phosphate Backbone with the Aid of "Chemical Nucleases"
Bonds in the sugar-phosphate backbone can be cleaved in the presence of O2 or H2O2, reducing agents, and chelated ions of transition metals. The oxidative degradation of nucleic acids was studied most thoroughly in experiments where used as chelating agents were some antibiotics, such as bleomycin (Blm) and its analogues, as well as ethylenediaminetetraacetic acid (EDTA), ortho-phenanthroline (Phen), and porphyrins.
Molecular oxygen is a four-electron oxidizing agent and its stepwise reduction to water can be written as follows:
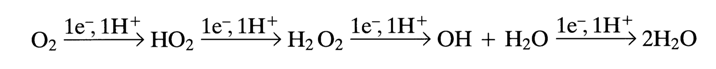
Molecular oxygen in an aqueous solution is a potent oxidizing agent, during reduction to both H20 and H2O2, the values of E0' being 0.815 and 0.3 V, respectively. However, direct oxidation with oxygen, whether a two- or four-electron one, fails to materialize. This is first of all due to spin forbiddenness, because the ground state of O2 is triplet. At the same time, one-electron reduction, which is the first step in the above scheme, is endothermic (E0 = -0.16 V). The role of the metal ion or complex boils down first of all to enabling this step. The resulting superoxide radical turns into H2O2 either by way of disproportionation or a reaction with a supplementary reducing agent H2A (ascorbic acid, thiols, etc.). Hydrogen peroxide is not capable of oxidizing most organic compounds at a measurable rate under mild conditions either, and its one-electron reduction also calls for a transition metal ion. Formed in this case are hydroxyl radicals that act as most potent agents (E0 = 1.9 V) capable of oxidizing any organic compounds. This process, just as the first step of oxygen reduction, also requires a conjugated easily oxidizable substrate H2A for reduction of the metal ion to the lowest state of oxidation. The active forms of oxygen emerging in the course of the process are in a free state as well as coordinated with the metal ion. By way of an example, here is the most probable scheme of oxidation of organic substrate H2X in the presence of metal ion or complex M+n and a conjugated reducing agent H2A (the oxygen particles are not coordinated):
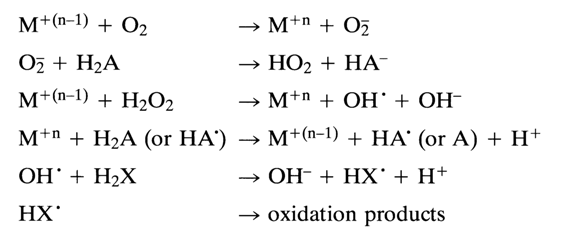
It can be seen that the metal complex acts as a reducing agent in reactions with 02 and H202 and, after subsequent regeneration, as an oxidizing agent. Consequently, the metal complex must have an intermediate value of the redox potential, which is to say that its behavior as a reducing or oxidizing agent must be moderate. Moreover, the ligands must readily lend themselves to substitution, because some of the electrontransfer reactions shown in the above scheme are intraspheric. These conditions are most fully met by copper complexes whose standard redox potentials are dependent on the ligand species but insignificantly and range from 0.1 to 0.2 V and which are highly labile with respect to substitution, as well as iron complexes, such as those with EDTA or porphyrins, whose redox potentials range from 0.15 to 0.30 V At the same time, the iron complex with ortho-phenanthroline cannot act as a catalyst because of the very high redox potential (E0 = 1.06 V). In principle, complexes of other transition metals, such as cobalt, nickel, and manganese, may also catalyze redox reactions involving nucleic acids.
Two pathways of degradation have been identified, different in composition of the end products. The first pathway leads to direct degradation of the chain with formation of 3-phosphoglycolates at the 5'end and a phosphate group at the 3' end, accompanied by liberation of propenal in the corresponding heterocycle. The second pathway leads to liberation of a heterocycle with the deoxyribophosphate backbone remaining intact but being prone to hydrolysis in a weakly alkaline medium. Such hydrolysis is typical of deoxyribophosphate fragments with a liberated heterocycle.
The mechanism of the process seems to involve detachment of the hydrogen at C4' during the limiting step. Direct degradation occurs when the emerging radical turns into a hydroperoxide one and then forms a hydroperoxide group. The latter undergoes rearrangement with the oxygen of the hydroperoxide group being inserted between C3' and C4'. As a result, the above-mentioned products of direct degradation are formed.
The main features of the mechanism of DNA cleavage with the aid of Fe(Ill)-EDTA derivatives were studied in an experiment with an ethidiumpropyl-EDTA-Fe(III) derivative. In this case, the 5'-terminal unit of the chain turned into an oligonucleotide with a 3'-terminal phosphate or phosphoglycolate, whereas the 3'-terminal unit carried a 5' -phosphate at the end. The process was accompanied by liberation of a heterocycle. The emerging three-carbon fragment was not identified. The degradation products were the same as during gamma radiolysis of DNA. This is indicative of direct involvement of free OH radicals at the first step of degradation.
Analysis of the products of DNA hydrolysis in the presence of a Cu(II)(Phen)2 complex has shown that the attack of the active oxygen-containing particle begins at C1' of deoxyribose. The hydrolysis products are fragments with 5'- and 3-phosphate groups, a free heterocycle, and 5-methylenefuranone. The differences in products of degradation by Cu(Phen)2 and Fe(111)-EDTA complexes indicate that the active oxygen-containing particles belong to different species.
This process results in cleavage of DNA with formation of 3'- and 5'-phosphorylated fragments at the site where the DNA chain is attacked. An alternative pathway comprises an attack of C4 in the nucleoside with liberation of a base and formation of a fragment with a 5'-phosphate group, while the second (5'-terminal) fragment has a substituted 3'-phosphate group.
Given below are the formulas of the chelate complexes of various metals most frequently used for degradation of DNAs under physiological conditions.
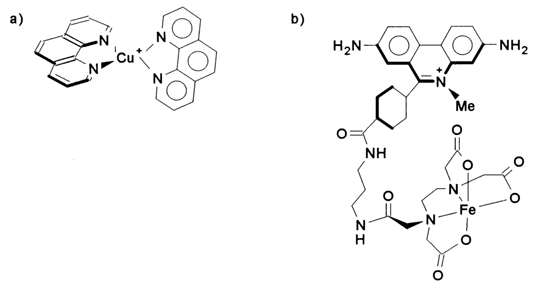
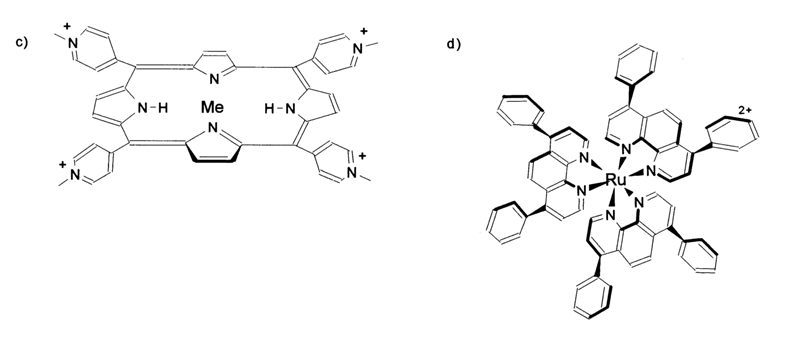
These and other similar compounds have become known as "chemical nucleases". They include, among others, the natural antibiotic bleomycin.
9.5.3 Reactions with Internucleotide Linkages Remaining Intact
Reactions of this type, involving internucleotide phosphate groups, have not been studied as thoroughly as those accompanied by cleavage of the polynucleotide chain. Only the substitution reaction has been described in this category.
Internucleotide phosphate groups in nucleic acids under normal conditions (aqueous medium, 200 C, pH ~ 7) exist in anionic form (pKa ~ 1) and, therefore, display nucleophilic behavior. This property is used in alkylation, whereby an internucleotide phosphate group is converted into a triester one. Along with alkylation of heterocyclic bases, conversion of this type involves attack of nucleic acids by such alkylating agents as nitrosoalkylurea. The latter is known as one of the most potent mutagenic compounds and as an antitumor drug. Studies of reactions between nitrosoethylurea and nucleic acids have demonstrated that this reagent is capable of alkylating internucleotide linkages (phosphodiester bonds) more effectively as compared to a reaction at heterocycles.
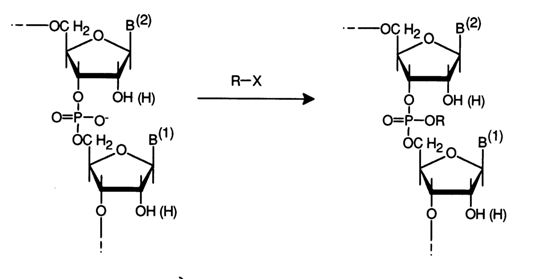
For instance, when viral RNA is modified with nitrosoethylurea by 70 %, the alkylation takes place at internucleotide phosphate groups. Carbamoylation occurs as a side reaction.
The phosphotriester internucleotide groups yielded by the reaction with nucleic acids are labile, especially in the case of RNAs. Such an alkylated RNA may be easily cleaved at alkylated internucleotide groups in a weakly alkaline medium. Intramolecular nucleophilic attack by the 2'-hydroxyl group ruptures internucleotide linkages.