![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
9.2.2 Modification of Heterocyclic Bases
As is known, the order in which heterocyclic bases alternate in nucleic acids as well as their nature exert a decisive influence on the functioning of nucleic acids, particularly, on the way they pass on genetic information. So when we examine the ways in which the chemical nature of pyrimidine and purine bases in nucleic acids, changes, that is, their chemical modification, we find ourselves face to face with the most exciting opportunity in the organic chemistry of nucleic acids - the possibility of tinkering with the structure of the genetic material.
The reactivity of pyrimidine and purine bases in nucleosides and nucleotides is not affected in any significant way by three-dimensional structure, and from the standpoint of chemical behavior they are similar to the corresponding free pyrimidines and purines (under conditions when the carbohydrate moiety or phosphate group is not involved). At the same time, as follows from the foregoing, the steric factors of the structure of nucleic acids significantly influence the reactivity of their constituent bases. Knowing these factors and also the nature of the reactive sites of the bases and the mechanisms of their reactions with various substances. one can select the right conditions for the modification to proceed in the easiest way along a definite route. This means that the reaction should not affect internucleotide linkages plus carbohydrate moieties and phosphate groups. Another implication is that the reagent's attack must be aimed at particular reactive sites of heterocyclic bases. This section deals successively with transformations involving each of the above discussed reactive sites. Hopefully, by the end of the chapter the reader will have a better understanding of how the structure of bases in nucleic acids can be modified in a controlled manner.
9.2.2.1 Reactions at Carbon Atoms
Interaction with Nucleophilic Reagents. The most reactive fragment in pyrimidine bases of nucleic acids, under conditions when the latter retain their three-dimensional structure (aqueous solutions, pH values close to neutral, moderate temperature), is the double bond C5=C6 deficient in electrons because of the effect exerted by the carbonyl groups.
The uracil and cytosine systems are in certain ways reminiscent of compounds in the ethylene series, containing an electron-acceptor substituent at the
a-carbon. Just as such compounds are attacked by nucleophilic reagents at the b-position, the nuclei of uracil and cytosine react with nucleophiles at C6. Since, as has already been pointed out, the nucleophile attacks the base at a right angle to the plane of the heterocyclic ring, such reactions are possible only if the base occupies a single-stranded segment of the nucleic acid. Stacking makes them impossible over double-stranded segments of RNA and DNA.It should be borne in mind that the interaction of nucleic acids with various reagents is much slower, as compared to nucleosides and nucleotides. Consequently, in order to ensure sufficiently fast reaction rates, nucleic acids are modified using reagents in excess amounts. As a result. the concentration of ions in the reaction mixture may reach a rather high level. In some cases, this disturbs the three-dimensional structure of the nucleic acid involved. This possibility must always be taken into consideration when nucleic acids are modified with a view to elucidating their spatial organization.
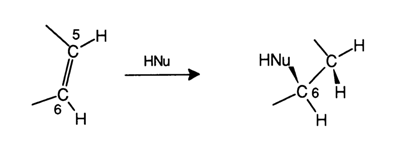
As a consequence of addition, the pyrimidine nucleus is not planar any more (it acquires the half-chair conformation similar to that in cyclohexene) and loses its aromaticity.
Introduction of alkyl substituents at C5 and C6 in the pyrimidine ring renders the latter significantly more stable toward nucleophiles. This is why thymines usually enter into such reactions with much greater difficulty. Purine bases are not readily involved in them either (the double C=C bond in the purine nucleus, corresponding to the C5=C6 bond in pyrimidines, is integrated into the aromatic system of imidazole).
Under the effect of nucleophilic agents, cytosines and adenines may undergo substitution of the exocyclic amino group. In the case of adenine, however, this reaction is extremely slow.
Thus, no acceptable methods for modifying purine bases in nucleic acids by nucleophiles have been devised so far.
For modification of uracils and cytosines use is made of O-methylhydroxylamine, sodium bisulfite and, less commonly hydrazine or alkylhydrazines. Depending on the reaction conditions and the reagents used, the initial addition at the double bond C5=C6 may be followed by both amino group substitution (for cytosine) and cleavage of the heterocyclic ring.
Let us, first, consider the addition and substitution reactions.
In the case of uracils, the addition of nucleophiles, such as hydroxylamine and bisulfite, proceeds at the fastest rate in a weakly alkaline medium (pH ~ 8) when the heterocyclic base is not ionized. O-Methylhydroxylamine does not enter into the reaction.
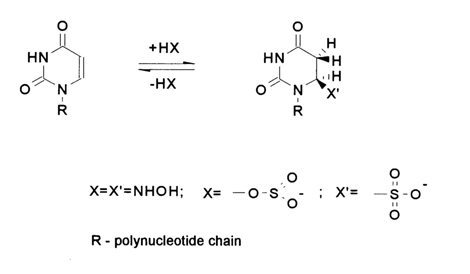
The resulting dihydrouracil derivatives are rather unstable and readily enter into a back reaction. The rate of hydroxylamine detachment is virtually independent of the pH of the medium, at pH values ranging from 0 to 6, but increases sharply with temperature (in an alkaline medium at pH
³ 10 the uracil nucleus breaks down). The bisulfite anion is easily detached in an alkaline medium at pH 9-10; the elimination reaction slows down at pH ³ 11. This indicates that dianion (1) does not participate in the reaction. There is evidence that the process takes the following course: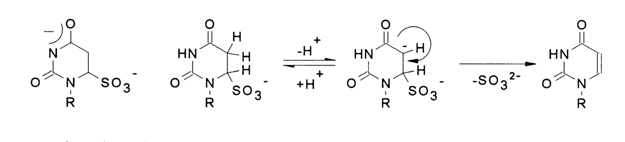
Consequently, the above reactions involving uracil derivatives cannot be used to obtain and subsequently investigate nucleic acids containing bases in a modified form; however, they must be taken into account in the calculations necessary for modifying nucleic acids by reagents.
The addition of such nucleophiles as hydroxylamine and its O-methyl ester as well as bisulfite at the double C5-C6 bond in cytosines proceeds easily at pH 5-7; that is, under conditions when the latter are partially protonated at N3. It is the protonated form that is the most active in this case.
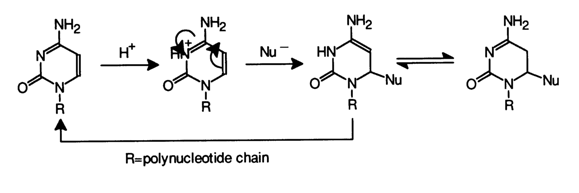
What sets the modification of cytosines apart is the fact that immediately after addition of the nucleophile at the C5-C6 bond (this reaction is reversible just as in the case of uracils) the amino group readily undergoes nucleophilic substitution. The following scheme summarizes all possible transformations affecting cytosines in the presence of nucleophilic reagents (HX and HY, see Table 9-1).
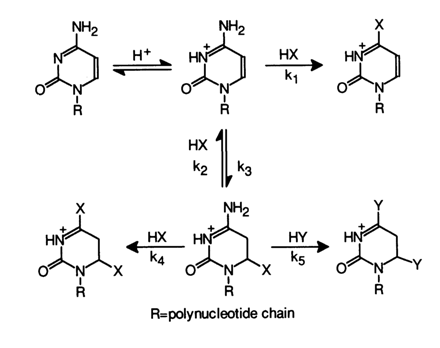
Addition at the double C5-C6 bond in the reaction with bisulfite anion proceeds at the fastest rate. If the reaction medium contains another nucleophilic reagent (HY), the amino group may be substituted by both nucleophilic groups X and Y The nucleophilic substitution in the aromatic ring of cytosine is rather slow (k1 << k2). Hydroxylamine, O-methylhydroxylamine, bisulfite ion, and some combinations of these reagents are usually the nucleophiles of choice for this type of nucleic acid modification (see Table 9-1). Of particular interest as a modification agent is the bisulfite ion. Its use makes it rather easy to attain an equilibrium which is markedly shifted toward adduct formation if the conditions are right (by more than 70 % in a 1 M solution of bisulfite at 200 C and pH 5). Under the same conditions, substitution of a hydroxyl for the amino group takes place at a rate slower than that of adduct formation (water in the above scheme acts as HY; k1 = k4 = 0, k5 < k2 and k3). The resulting deamination product is similar to the adduct of uracil and bisulfite and easily loses the bisulfite anion at pH 8-9 to turn into a respective uracil derivative. Knowing the optimal pH value for each step of this process, one can convert nucleic acid cytosines into uracils under sufficiently mild conditions. This method of nucleic acid modification can be used, for example, in secondary structure studies.
It should be borne in mind that if the reaction with bisulfite is conducted at pH 7, virtually no cytosines are converted into uracils.
A rather important application has been found for a reaction between cytosines and bisulfite in the presence of nucleophiles more active than water. Such nucleophiles have usually been various amino compounds of the NH2R.
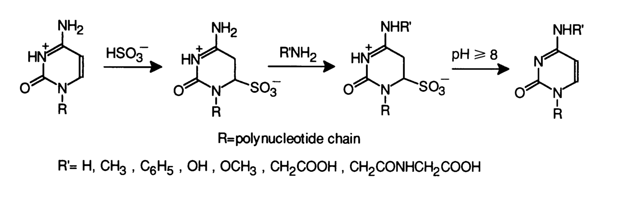
The optimal pH value for such a modification of cytosines depends, naturally, on pKa of the amino compound used (the latter acts as a nucleophile when the amino group is not protonated).
Thus, in the case of cytosines the above reactions can, according to the end results, be classified as nucleophilic substitution at C4.
Table 9-1 summarizes reactions between cytosines of nucleic acids and nucleophiles, proceeding at C4 (k1, k4, k5) and C6 (k2).
As has already been mentioned, addition of nucleophiles at the double C5=C6 bond may in certain instances be followed by rupture of the heterocyclic rings in pyrimidines. Such transformations involving uracils, thymines and cytosines have been observed while treating nucleic acids with hydrazine at pH > 8.
Table 9-1. Reaction of the Cytosine Fragment in Nucleic Acids with Nucleophilic Reagents " + " - the reaction proceeds; " - " - the reaction does not proceed.

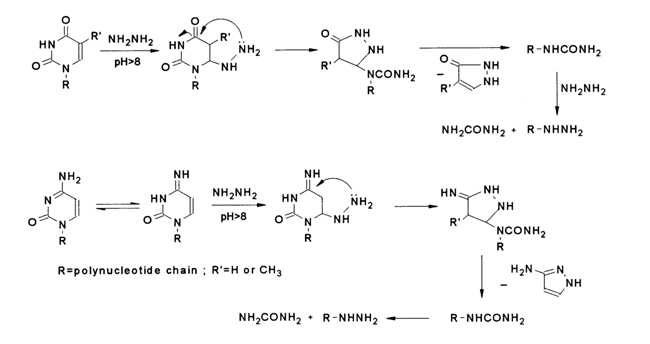
For removal of pyrimidine bases nucleic acids are normally treated with anhydrous hydrazine (600 C, 20 h) to avoid cleavage of phosphodiester bonds in an alkaline medium. A similar result can be achieved when aqueous solutions of hydrazine are used at controlled pH values (pH 9.5, 00 C). Hydrazine treatment of DNA leads to complete elimination of cytosines and thymines. As regards RNA with its more reactive uracils (in comparison with thymines), complete elimination of pyrimidines is much faster. When, for example, tRNA is treated with anhydrous hydrazine (370 C, 15 h), all uracils and cytosines are eliminated. A side process of note is cleavage of internucleotide linkages or, in other words, depolymerization.
The residual hydrazine can be eliminated from nucleic acids depyrimidinated in this fashion with the aid of aldehydes (ketones) so that the site previously occupied by the pyrimidine unit in the nucleic acid now accommodates heterocycle-free ribose whose phosphodiester bonds are highly labile and can break under mild conditions. This procedure is widely used to break nucleic acids down in determining their primary structure.
Hydroxylamine acts on nucleic acids in an alkaline medium similarly to hydrazine but in a more specific manner. At pH
³ 10 it breaks down only uracils. This reaction resembles that of cytosines and uracils with hydrazine under identical conditions.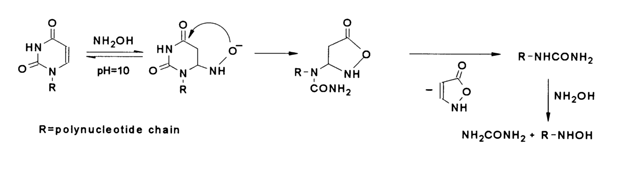
The specificity of this reaction makes it possible to selectively modify uracil nuclei in RNA. The optimal conditions for the reaction are as follows: pH 10, 10 M solution of hydroxylamine, 100 C, 150 h. The last step boils down to substitution of hydroxylamine for urea in the corresponding segments of the polynucleotide chain. Under the above optimal conditions, the nonspecific modification of the polynucleotide chain is minimum.
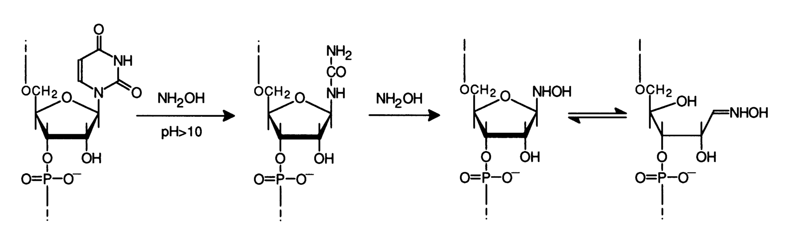
Just as hydrazinolysis, the reaction of DNA and RNA with hydroxylamine is employed for controlled cleavage at base-free units. The reaction with hydroxylamine is sensitive to the secondary nucleic acid structure (under the reaction conditions the secondary structure seems to persist) so that only uracils outside double-stranded segments are cleaved. In the case of tRNA, for instance, only those uridine units undergo modification under normal conditions which are located in loops, whereas at 370 C or at a low temperature but in the presence of urea which destroys the secondary structure uracils are cleaved completely.
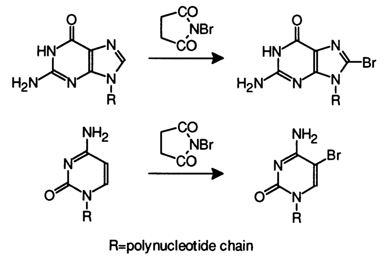
Interaction with Electrophilic Reagents. As is known, electrophilic substitution in pyrimidine bases forming part of nucleotides occurs at C5, While in purine bases it occures at C8. These bases behave similarly in nucleic acids as well. This section deals only with reactions taking place in water under mild conditions favorable for nucleic acids to retain their three-dimensional structure.
These conditions are ideally provided by halogenation and mercuration. The halogenation reaction involves uracil, cytosine, thymine, and guanine. Just as in the cases described above, the halogenating agent attacks the reactive site at an angle normal to the plane of the heterocyclic ring, which is why the secondary structure and other structural features of nucleic acids impose the same limitations on the reactivity of the constituent bases as the use of nucleophilic reagents.
The bromination of uracil and cytosine in nucleic acids can be achieved by adding bromine to water at pH values close to neutral.
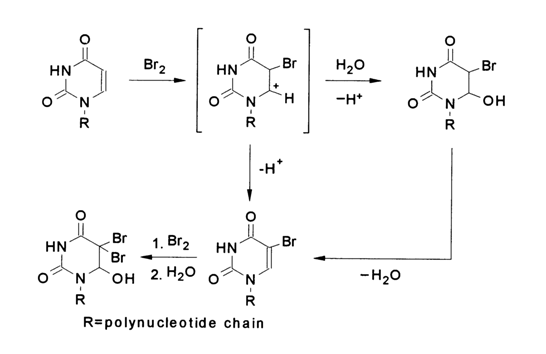
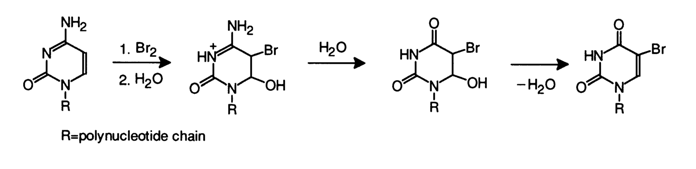
In nucleic acid cytosines whose modification also proceeds according to the above scheme, the bromohydrin which is formed first easily undergoes substitution of a hydroxyl for the amino group (just as the adduct of cytosine with bisulfite) with subsequent release of water to give 5-bromouracil:
The modification of nucleic acid thymines comes to a halt at the step of formation of the respective bromohydrin.
Treatment of nucleic acids with bromosuccinimide modifies cytosines and guanines, the modification of the former occurring predominantly at pH 9 and that of the latter at pH 7.
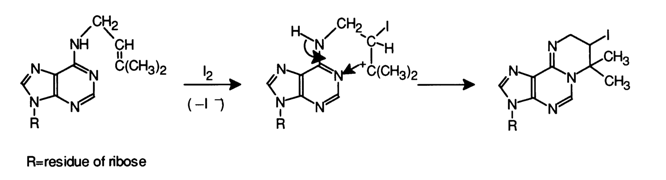
To incorporate iodine into polynucleotides ICI or I- is normally used in the presence of TlC13 (mild oxidizing agent) because free iodine is not reactive enough. Iodine itself, however, is a specific agent with respect to some tRNAs because some constituent minor bases of the latter do enter into a reaction with it. For example, the reaction with 6-isopentenyladenosine yields a new cyclic system:
When iodine chloride is employed as the iodination agent, the halogenation proceeds in the same fashion as bromination. The wide use of iodination to modify nucleic acids stems from the fact that this technique allows relatively stable isotopes (the half-life of 125I, for example, is about 25 days) to be incorporated into them under mild conditions, which enables one to use radiometric methods and handle nucleic acids in ultramicroamounts. Since incorporation of an iodine isotope does not alter the specific interactions in nucleic acids (three-dimensional structure), this approach is highly promising for structural and functional studies.
Another reaction of electrophilic substitution - mercuration - is also used for modification of nucleic acids apart from halogenation. The mercurating agent, which is usually mercuric acetate, reacts with pyrimidine bases at position 5 without affecting purines and thymines.
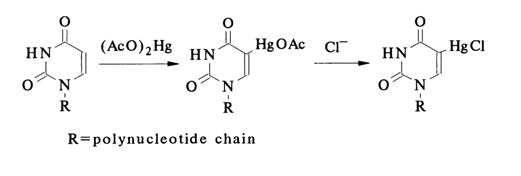
Uracils and cytosines, which react in a similar manner, are converted into the corresponding 5-mercury substituents as part of both mononucleotides and nucleic acids. The reaction proceeds in buffer solutions at pH 6-7. The covalent (or, to be more precise, coordination covalent) C-Hg bond is rather strong in such compounds under conditions usually maintained while conducting biochemical reactions. Treatment of nucleic acids with excess mercuric acetate leads to substitution for the hydrogens at C5 of all pyrimidines. It should be remembered that the course taken by mercuration of nucleic acids is independent of their secondary structure, although this reaction must proceed, similarly to halogenation, as an attack perpendicular to the base plane. This may stem from the fact that the ion Hg 2+ with its pronounced capacity for complexing is retained, at first, by the nitrogens of the heterocyclic bases through noncovalent interactions and then gets sandwiched (intercalates) between base planes which are involved in stacking. When a limited number of bases are to be modified, the reagent is used in a limited amount. Any RNAs and DNAs (including synthetic polynucleotides) may enter into the reaction.
Mercurated polynucleotides (even completely mercurated DNAs and RNAs) retain their capacity for complementary interactions (the HgX moiety lies in the major groove) and may serve as templates and primers in certain nucleic acid biosynthesis reactions involving polymerases.
Mercurated double-stranded DNAs are characterized virtually by the same melting points and display the same kind of hypochromism as the starting DNA molecules.
Mercurated nucleotides and polynucleotides readily react with mercaptans, which becomes useful in immobilization of polynucleotides on sulfhydryl Sepharose
?:
This reaction, yielding a rather stable C5-Hg-S group, shows a great deal of specificity and proceeds at a high rate: the immobilization is rather easy even for preparations with only one mercury atom per 200 nucleotides. Elution with a buffer containing mercaptan (mercaptoethanol, etc.) removes the polynucleotide from Sepharose (as a result of exchange). This technique for immobilizing polynucleotides on and removing them from the carrier under very mild conditions is employed to separate and isolate mercurated polynucleotides from complex mixtures where the latter are used as templates or primers in biosynthesis of DNAs and RNAs.
Mercurated nucleotides rather easily exchange a mercury atom for a halogen or tritium if the corresponding compound is treated with such agents as iodine, N-bromosuccinimide, sodium borohydride, and so on. For instance:
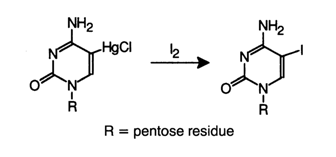
Polynucleotides can easily exchange a mercury atom as well, which is used when isotopes of iodine (reagent 125I2) or tritium (reagent NaBT4) are to be incorporated into DNA and RNA molecules. Such a labeling method is of particular importance for RNAs because the corresponding forward reactions involving the latter are accompanied either by formation of side products (e.g., dihydrouracil derivatives) or cleavage of internucleotide linkages (when the exchange takes place in an alkaline solution of T2O).
The mercuration of RNAs and DNAs as well as synthetic oligo- and polynucleotides may be used in studying the nucleic acid structure by electron microscopy and other techniques.
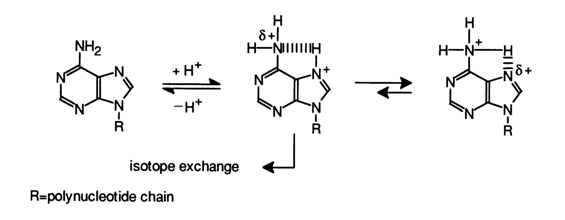
Isotopic Exchange of Hydrogen. Polynucleotides are typically involved in isotopic exchange of hydrogen atoms in aqueous media containing deuterium and tritium. Distinction is made between "fast" and "slow" isotopic exchange. During fast isotopic exchange, the state of equilibrium is attained within seconds or a few minutes even at 00 C. Groups participating in fast isotopic exchange include the phosphates and hydroxyls of the carbohydrate-phosphate backbone as well as the amino and imide groups of heterocyclic bases. The high rates of exchange observed in all cases are due to the high rates of protonation and deprotonation of oxygen and nitrogen atoms. Thereby, any alteration in the electronic state or accessibility of oxygen or nitrogen (e.g., when exocyclic amino and other groups as well as heterocycles are involved in complementary pairing) is followed by a drastic change in the isotopic exchange rate.
Analysis of the kinetic data pertinent to isotopic equilibrium makes it possible to identify rather clearly defined groups with characteristic isotopic change rates. Such analysis permits one, for example. to estimate the involvement of particular nucleic acid bases in complementary interactions, to determine the relative number of single- and double-stranded segments in RNAs and DNAs, and to monitor changes in the intramolecular organization of nucleic acids and nucleoproteins.
By slow exchange in nucleic acids is meant the reaction of isotopic exchange of the hydrogens bound to carbons of heterocyclic bases. Such an exchange is observed when a nucleic acid is heated for many hours in deuteriated or tritiated water at elevated (about 1000 C) temperatures. It has been found that only the hydrogen at C8 in purines and C5 in pyrimidines is capable of entering into the isotopic exchange reaction. The hydrogen at C6 in pyrimidines also tends to enter into such a reaction but at an extremely slow rate and requires rather rigorous conditions. The rest of the hydrogens bound to carbons do not enter into isotopic exchange reactions in aqueous solutions 1).
The slow isotopic exchange in purines is essentially a first-order reaction and proceeds at a rate 30 to 100 times faster than in pyrimidines. The so-called ilide mechanism has been proposed to account for the isotopic exchange in purine derivatives. According to this mechanism, the following sequence of reactions leading to the isotopic exchange can be written for guanines in a polynucleotide chain:
1)
When dry nucleic acid preparations are irradiated with tritium or deuterium atoms that is, under conditions of heterogeneous exchange - all hydrogens in nucleic acids may enter into isotopic exchange reactions. The fastest exchange involves the methyl hydrogens in thymines.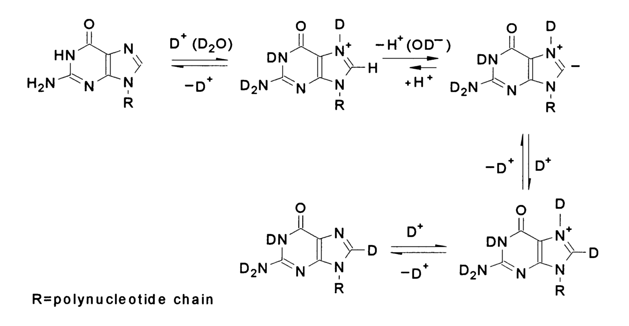
The first step of the process is protonation of the imidazole ring at N7 This is one of the two steps limiting the entire process (the protonation itself is rather fast but the equilibrium concentration of the protonated form which continues to react is small). The second, slow step is proton detachment from C8, mediated by a hydroxyl ion, to form an ilide. The resulting carbanion abstracts a proton (deuterium or tritium) from water, which eventually leads to isotopic exchange at C8.
A similar mechanism of isotopic exchange of hydrogen at a respective carbon has been demonstrated for imidazole and related five-membered heterocyclic systems.
Interestingly, favorable conditions for the first limiting step are created in an acid medium, while those for the second step are created in an alkaline one. This results in a rather complex relationship between the isotopic exchange rate and pH. In the case of guanines, for instance, an increase in pH from 4 to 8 affects only insignificantly the rate of isotopic exchange but at pH ranging from 8 to 11 the reaction rate increases sharply, as much as almost ten-fold. A further increase in the concentration of hydroxyl ions up to 1 N does not bring about any significant changes in the isotopic exchange rate.
As regards adenines, the isotopic exchange in neutral and weakly alkaline solutions is based on the same mechanism but proceeds at a perceptibly slower rate than in the case of guanines, which may be explained by the diminishing positive charge at N7
due to intramolecular protonation of the exocyclic amino group.The rate of exchange in adenines varies slightly in the pH range of 4 to 12, but in concentrated alkalies (> 0.1 N) it increases drastically in proportion to the hydroxyl ion concentration. The mechanism of this phenomenon is yet to be elucidated.
Purine derivatives carrying a positive charge at N7 (e.g., N7-methylguanine, Nl, N7-dimethylguanine, N7-methylinosine) have been found to display rates of the isotopic exchange at C8 comparable to those of fast isotopic exchange. This principle underlies an original method for determining the degree of methylation of guanines in DNAs. Taken for the test is a 3H-DNA sample
with tritium atoms at C8 of guanines, which is then treated with dimethyl sulfate, and the percentage of the tritium capable of rapid exchange with water is determined as a measure of modification (methylation) of N1 of guanine in the DNA.
As has already been mentioned, the rate of the slow isotopic exchange at C5 in pyrimidines is much lower than in purines. This exchange, however, is accelerated significantly in the presence of compounds capable of undergoing reversible addition at the double C5=C6 bond. Such compounds may be citrates, bisulfite ion, 2-mercaptoethylamine, ethylamine, ethanolamine, cysteine, and so on. In this case, the isotopic exchange rate becomes proportional to the concentration of the corresponding homogeneous catalyst and may be tens and hundreds of times higher. The mechanism of this homogeneous catalysis resides in reversible addition of respective compounds at the C5 = C6 bond in pyrimidines. The exchange in a cytosine with bisulfite ion serving as the catalyst can be written as follows:
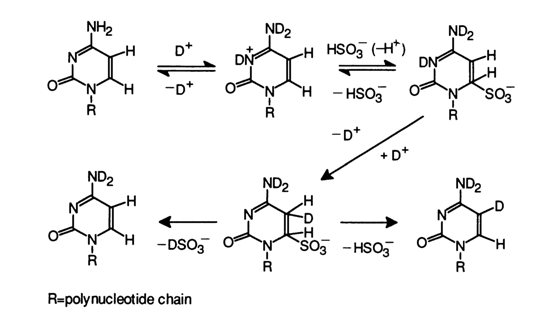
It can be seen that the addition proceeds in two steps: 1,4-addition to a conjugated system of double bonds (N3, C6), followed by intra- or intermolecular transfer of the isotope from N3 to C5 . The last step is not sterochemically directed so that the deuterium added to C5 may find itself either in syn or anti position with respect to the remaining hydrogen atom (remember that the emerging hydrogenated pyrimidine ring is not planar). The subsequent
b-elimination seems to proceed in a single step and in a stereochemically directed manner with removal of the bisulfite anion and hydrogen that are either in anti-anti or syn-syn positions (trans-detachment). Thus, during regeneration of the starting cytosine some hydrogens become substituted by deuterium. When the addition-detachment cycle is repeated over and over, isotopic equilibrium is achieved, which is to say that we are dealing with an isotopic exchange reaction.A similar mechanism underlies exchange in uracils as well, with the difference that the exocyclic oxygen at C4 is involved in the 1,4-addition.
Reversible addition of some nucleophiles at the double C5=C6 bond has also been described for thymines; however, it must not (and does not) lead to isotopic exchange of hydrogen at carbon atoms.
Interestingly, the best catalysts of isotopic exchange at C5 in pyrimidines are bifunctional reagents of the cysteine or 2-mercaptoethylamine type. The catalysis of isotopic exchange in uracil by deuteriated 2-mercaptoethylamine can be represented as follows:
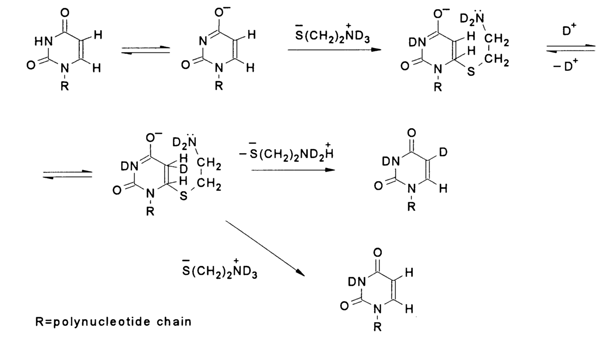
It is evident that in the case of bifunctional agents of the 2-mercaptoethylamine type, where the mercapto group acts as a nucleophile and the amino group acts as a base, intramolecular catalysis of detachment of the added nucleophile is possible, which increases the rate of addition-detachment or, in other words, isotopic exchange.
The different optimal pH values for these reactions, depending on whether they involve cytosines (pH 5-7) or uracils (pH 7-9), make it possible to conduct isotopic exchange selectively for each of the two bases in the heteropolynucleotide. Moreover, it should be pointed out that the reaction of isotopic exchange at C5 in pyrimidine derivatives is contingent on possible attack of C6 by a nucleophile at a right angle to the ring plane. The reaction is virtually at a standstill in native double-stranded DNAs and double-stranded segments of single-stranded DNAs and RNAs where such an attack is hindered. This property is often put to practical use when it is necessary to determine the involvement of a particular pyrimidine in the formation of secondary RNA or DNA structure.
The slow isotopic exchange of hydrogen at C6 in pyrimidines, which may occur without any specific catalyst as well, is based on the same addition-detachment mechanism with water acting as a nucleophile and proton donor.
The slow isotopic exchange reactions described in this section are widely used in preparing tritium-labeled DNA and RNA. To this end, a nucleic acid preparation is kept at high temperature in tritiated water till isotopic equilibrium is reached and the rapidly exchangeable tritium is eliminated. The nucleic acid isolated after such treatment contains only slowly exchangeable tritium (tritium atoms at restrictive positions). Being easy to prepare, nucleic acids labeled with tritium in this fashion do not undergo any marked degradation and modification, whereas the loss of tritium in aqueous solutions (pH 3-10) is negligible due to slow reverse isotopic exchange.