![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
9.2.2.2 Reactions at Pyridine Nitrogens
As a consequence of the presence of lone-pair electrons not participating in the formation of an aromatic sextet and the lack of bonds with hydrogen atoms, the pyridine nitrogens acting as reactive sites in nucleic acid bases react primarily with electrophiles. The most commonly used procedures for modification at these atoms are alkylation, production of N-oxides, and the reaction with diethylpyrocarbonate.
Alkylation. In assessing the possibilities of alkylating heterocyclic bases in nucleic acids, one must first of all compare the nucleophilic properties of the reactive sites in their molecules. It has already been mentioned that such properties must be most pronounced at pyridine nitrogens. In order to determine which of them will be alkylated most readily (exhibit nucleophilic properties by giving up a pair of electrons for binding with a carbon) and in what bases, it is advisable to bear their capacity for protonation in mind (i.e., capacity to give up an electron pair for binding with a hydrogen, thereby displaying basic properties), since it is known that the nucleophilic and basic behavior of nitrogen atoms in organic compounds varies concomitantly. As has been pointed out, cytosines, adenines and guanines in nucleosides and nucleotides are protonated, respectively, at N3, N1 and N7 (the reasons why the exocyclic amino groups in cytosines and adenines do not lend themselves to protonation have already been discussed), whereas uracils and thymines do not exhibit any basic properties at all. However, the differences in the behavior of adenines and guanines during protonation are yet to be explained. The only assumption that can be made is that the lower basicity of N7 in adenines, as compared to guanines, has to do with screening by the exocyclic amino group.
According to the ease with which the above bases enter into the
reaction, they can be arranged in the following order:
guanine (N7) > adenine (N1) > cytosine (N3). The involvement of other pyridine
nitrogens is much more insignificant.
Just as with other types of nucleic acid modification, alkylation is usually conducted in aqueous solutions at neutral or slightly acid pH values. As the medium becomes more acidic (pH < 6), when the bases are predominantly in a protonated form, alkylation naturally comes to a halt.
At alkaline pH values. - NH - C = O groups in uracils thymines and guanines lose their proton to pass into the anionic form - N = C - O - which, as has already been mentioned, is alkylated at the nitrogen atom. For instance, the alkylation of uracils and thymines at alkaline pH values can be represented as follows:
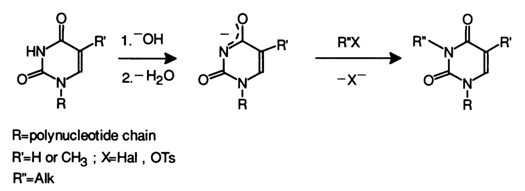
Similarly, at pH > 8, guanines are alkylated at N1 (along with formation of an N7-alkyl derivative).
Under normal alkylation conditions (pH 6-7, aqueous solutions, 20-370 C), the alkylating agent attacks the heterocyclic pyridine nitrogen in the base plane. This is why stacking does not interfere with the reaction and the modification may involve bases forming part of double-stranded segments of nucleic acids. In this case, however, attack of these nitrogens by the alkylating agent is impossible (unless the double helix is damaged) because N1 of adenine and N3 of cytosine participate in hydrogen bonding. Therefore, when native DNAs are alkylated, the modification must occur primarily at N7 of the guanine located in the wide groove and, to a lesser extent, at the N3 of adenine. The alkylation of RNAs involves guanines, adenines and cytosines in single-stranded segments and only guanines in double-stranded ones. Since this method allows the investigator to find differences in the three-dimensional structure of nucleic acids, it is considered to hold a certain degree of promise for structural studies. More particularly, such an approach can be used in studying the structure of nucleoproteins where failure of guanines to be alkylated may suggest that a given portion of the nucleic acid is blocked by a protein. Another area of application of alkylation reactions is selective rupture of internucleotide linkages in nucleic acids. Since the glycosidic bond in an alkylated guanosine (which, as has been pointed out, is alkylated at a maximum rate) can be cleaved rather easily, one can produce polymers without guanines at selected points.

The same applies to segments with adenines alkylated at N3. Such selective cleavage of the polynucleotide chain forms the basis of a new chemical method for determining the nucleotide sequence in DNAs.
The most frequently employed alkylating agents include alkyl halides, dimethyl sulfate, methyl methanesulfonate, and diazomethane. Analogues of nitrogen mustards, such as N-aryl-p-chloroethylamines, are also used for the purpose. The alkylation with alkyl halides as well as sulfates and sulfonates is based on the usual mechanisms of nucleophilic substitution.
Which bases are methylated in the presence of diazomethane depends, as has already been indicated, on the reaction conditions: in the absence of proton donors it is pyrrole nitrogens that are methylated, whereas with protonation pyridine nitrogens are involved. It should be noted that the methylation of polynucleotides with diazomethane is always accompanied by cleavage of phosphodiester bonds (i.e., degradation of the polymer chain) and methylation at the 2'-hydroxyl groups of carbohydrate moieties (if such are present). At the polymer level this reaction is used when it becomes necessary to methylate uracils or thymines; it should be remembered, though, that the reaction is not selective - it also involves guanines as well as, to a lesser extent. cytosines and adenines.
Alkylation with N-arylchloroethylamines initially yields an imonium cation which then reacts with the base acting as the nucleophile Nu.
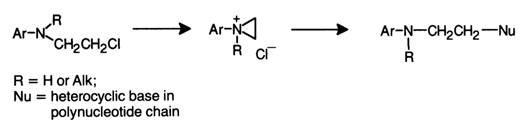
In this case, steric factors must have a more tangible effect on the course of alkylation, as compared to methylation, by virtue of the larger size of the electrophilic particle (imonium cation).
Production of N-Oxides. Pyridine nitrogens in some bases (of nucleic acids) are easily oxidized with peracids to yield N-oxides. The mechanism of this reaction is similar to that of alkylation because added to the atomic oxygen generated by the peracid is a free electron pair of the nitrogen being attacked (i.e., the attack proceeds in the plane of the heterocyclic nucleus).
Nucleic acids are usually modified by means of monoperphthalic acid which is one of the most stable peracids. The reaction is conducted in aqueous solutions at neutral pH values and at temperatures ranging from 0 to 200 C. In the case of oligonucleotides, whose reactivity is not affected by steric factors in any significant manner, adenines react with particular ease under such conditions to yield an N1-oxide. Cytosines react at a much slower rate and yield an N3-oxide. It should be borne in mind that the reactivity of adenines and cytosines in a nucleic acid may vary substantially. Guanines and uracils (as well as thymines) do not react under the above conditions.
Since, as has already been mentioned, the nitrogen atom is attacked during oxidation in the ring plane, this reaction affects only single-stranded segments of nucleic acids. As regards double-stranded segments, the oxidation of N1 in adenines and N3 in cytosines with oxygen is inhibited practically a hundred per cent because these atoms are involved in hydrogen bonding.
Reaction with Diethylpyrocarbonate. Diethylpyrocarbonate is a rather important reagent used for modifying heterocyclic bases in nucleic acids. As it reacts with nucleophiles, the compound yields carbethoxylation products, the ethoxycarbonate anion acting as the departing group. This anion easily breaks down in an aqueous solution, whereas the resulting carbethoxy derivative is rather stable toward hydrolysis. N-Carbethoxyimidazole, for example, is a hundred times more stable than N-acetylimidazole.
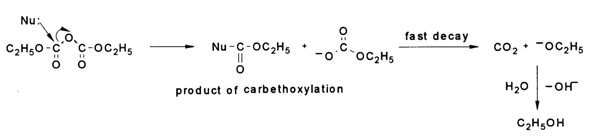
Thus, if the pyridine nitrogen of a purine or pyrimidine base acts as a nucleophile, diethylpyrocarbonate attacks it in the base plane to yield an amide-type derivative in which the positively charged nitrogen forms a rather strong linkage with the carbethoxy group; at the same time, the rapid degradation of the departing group will lead to a situation in which the hydroxyl anion becomes a counterion with respect to the resulting ammonium cation. For instance:
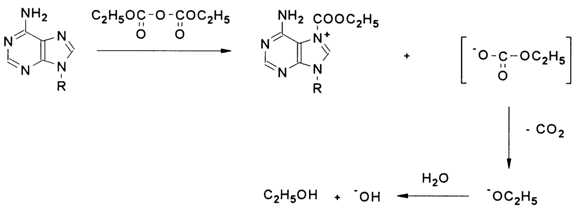
Being a strong nucleophile, the hydroxyl anion immediately attacks the neighboring C8 atom. In this case, the attack is perpendicular to the heterocyclic base plane. The subsequent transformations result in opening of the imidazole ring (R - polynucleotide chain):
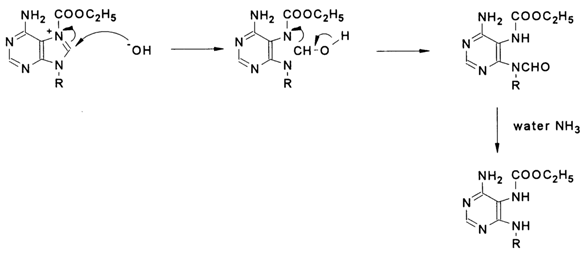
When the reagent is present in excess and the reaction takes a sufficiently long period of time, the exocyclic amino group is acylated as well.
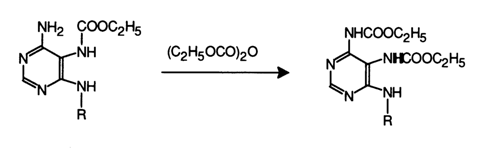
Guanines react similarly, but at a slower rate, with the difference that the intermediate formyl derivative is so unstable that it is immediately hydrolysed by water:
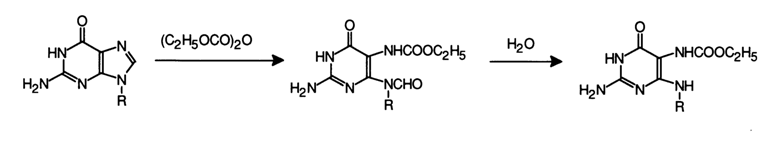
Cytosines are acylated with diethylpyrocarbonate at the exocyclic amino group and only if the reagent is used in an amply excess amount. Uracils and thymines do not get modified by diethylpyrocarbonate.
Since diethylpyrocarbonate is moderately soluble in water and easily hydrolysable, it is normally used as an emulsion and in a markedly excess amount. The reaction is carried out in neutral or almost neutral media and at room temperature.
Since, as has already been pointed out, the base is attacked during the reaction not only in the heterocycle plane (the first step of modification of adenines and guanines is acylation of N7 atoms), but also at a right angle thereto (the second step of modification of adenines and guanines is nucleophilic addition of the hydroxyl anion at C8 plus acylation of the exocyclic amino groups in adenines and cytosines), diethylpyrocarbonate can be used to modify only single-stranded segments of nucleic acids. Consequently, the above reactions may involve oligonucleotides, denatured DNAs and RNAs. As a rule, more complex molecules enter into a reaction with diethylpyrocarbonate more sluggishly. For example, if the modification of a dinucleotide with adenines is a matter of just a few minutes, that of an RNA takes more than three hours to produce the same results. Since the reaction of adenines and guanines with diethylpyrocarbonate is accompanied by opening of the purine ring, it can be easily monitored by decreasing intensity of the UV absorption maximum typical of purine derivatives (259 nm in the case of adenine derivatives).
Studies into the behavior of homopolyribonucleotides in this reaction have shown that poly(U) does not enter into the latter, while poly(A), poly(G) and poly(C) are modified. Under conditions when the secondary structure cannot be upset, each base in poly(A) is involved in the reaction, whereas in poly(G) and poly(C) this is true only for one out of three and one out of five bases, respectively. Such a difference in behavior is due not only to the lower reactivity of guanines and cytosines, as compared to adenines, but also to the fact in poly(G) and poly(C) some bases participate in hydrogen bonding. The latter assumption is borne out by evidence showing that the poly(U) . poly(A) complex does not react with diethylpyrocarbonate at all.