![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
8.2 RNA
8.2.1 Secondary Structure of RNA. Fresco-Alberts-Doty Model
As had been established by the late fifties, in contrast to DNA, macromolecules of the overwhelming majority of cellular and viral RNA are single-stranded. None the less, in solutions of sufficiently high ionic strength RNA exhibited pronounced Cotton effects as well as hyperchromicity in the UV region. This was unambiguous evidence that RNA has an ordered secondary structure.
At the same time, the melting curves of RNA differed markedly from those of DNA and other complementary double-stranded polynucleotides. The transition of RNA from ordered to denatured state occurred in a rather broad temperature range (Fig. 8-23). What is more, it was found that RNAs in an ordered state (at "physiological" temperatures and salt concentrations) have a compact tertiary structure approaching that of the Gaussian coil. This was construed as evidence of presence of flexible single-stranded segments in RNA molecules. Notably, denaturation of ordered components of the RNA macromolecule is also followed by breakdown of this compact structure.
In 1960 such experimental evidence lead J. Fresco, B. Alberts and P. Doty to propose a model of secondary structure of single-stranded RNAs (Fig. 8-24). According to the model, the secondary structure of a single-stranded RNA is a combination of double-helical segments (widely differing in length and composition) and interlinking single-stranded ones. Thus, it was postulated that double-helical RNA segments result from base pairing in complementary nucleotide sequences belonging to the same strand. Fresco and coworkers hypothesized that this pairing obeys the Watson and Crick rule or, in other words, canonical G . C and A . U pairs are formed. The polynucleotide chain segments forming double helices are antiparallel. At one end or, more precisely, where the polynucleotide chain of RNA folds back on itself, the paired regions are connected by single-stranded hairpin loops of different length. Fresco and coworkers assumed that the double-stranded segments of RNA are not perfect double helices but contain internal loops and all kinds of bulges.
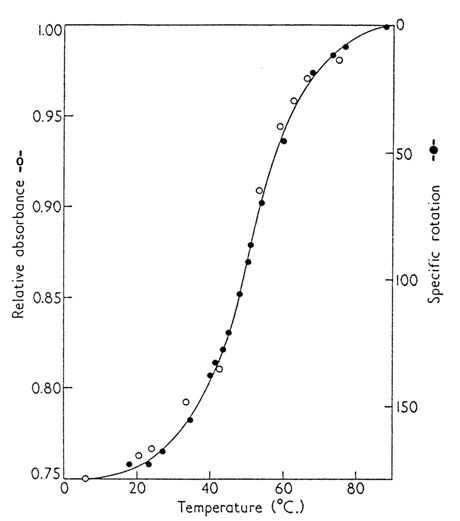
Fig. 8-23. Typical melting curve of single-stranded RNA (from tobacco mosaic virus) in 0.1 M phosphate buffer, pH 7.0 (adapted from P. Doty et al., Proc. Natl.Acad. Sci. USA, 45, 482-499 (1959)).
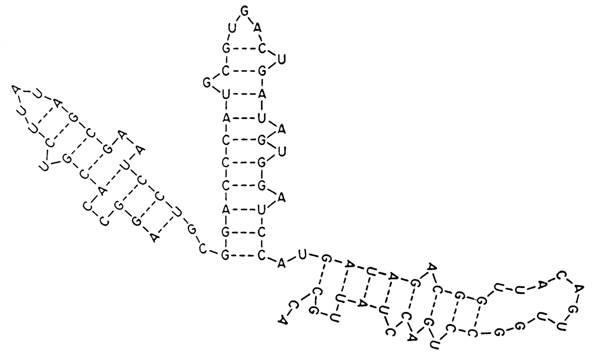
Fig. 8-24. Fresco-Alberts-Doty model of the secondary stucture of a single-stranded RNA (reproduced with permission from J. Fresco, B. Alberts and P. Doty, Nature, 188, 98-101 (1960)).
All subsequent studies into the macromolecular structure of RNA supported the Fresco-Alberts-Doty model, and it may be considered as a universal description of the secondary structure of single-stranded RNAs. At the same time, the model was expanded by addition of significant details. It has turned out that in addition to classical Watson-Crick pairs double-stranded segments of RNA often contain G . U pairs whose structure was discussed above. Formation of other noncanonical pairs has also been assumed. Moreover, as was demonstrated for many RNAs, double-stranded segments may result from pairing of not only neighboring complementary segments but also sequences spaced rather far apart in the polynucleotide chain. Consequently, double-stranded segments of RNA form branches of different kinds. All these features of the secondary structure can be observed in fragment 23S of ribosomal RNA, illustrated in Figure 8-25.
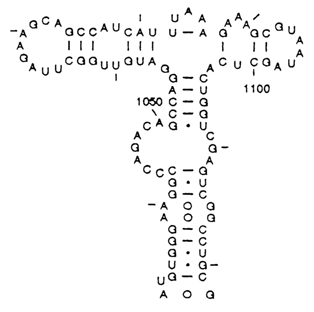
Fig. 8-25. Secondary structure of the segment of the E. coli 23S ribosomal RNA (adapted from R. R. Gutell et al., Nucl. Acid Res., 20, Supplement, 2095-2109, (1992)).
8.2.2 Elements of Secondary Structure
Let us now consider at greater length the properties of the structural elements constituting RNA macromolecules. Most of information on the subject is provided by the three-dimensional structure of transfer RNAs (tRNAs) as well as the results of studies into the conformation and thermodynamic stability of synthetic oligoribonucleotides which serve as models of a particular secondary structure motif of RNA.
Transfer RNAs were the first nucleic acids whose nucleotide sequences had been established. Moreover, several representatives of tRNA had been crystallized and investigated by high-resolution X-ray structural analysis. The results will be discussed in greater detail in 8.2.3. Here it should simply be pointed out that the secondary structure of any tRNA regardless of its nucleotide sequence is consistent with the "clover-leaf" model (Fig. 8-26). It can be seen that in essence the clover-leaf structure is quite compatible with the overall Fresco-Alberts-Doty model of RNA secondary structure.
Base-paired Regions. X-ray structural analysis of double helices of RNA has shown that they belong to the A family (i. e., are similar to A-DNA in many respects). Indeed, complementary base pairs in double-stranded segments of RNA lie on the periphery of the helix (Fig. 8-27) and the base planes are significantly (almost by 150) deflected from the axis of the helix. Two forms of RNA helices are known: form A with 11 base pairs per turn and form A' with 12 base pairs per turn. Form A may be converted into form A' if the salt (NaCl or KCl) concentration in the sample goes up.
As has already been mentioned, a characteristic structural feature of the A family of double helices of nucleic acids is the C3'-endo-conformation of the sugar. Insofar as helical RNA is concerned, such a conformation of the ribose seems to be the only possible one and, consequently, transition A ® B for this class of nucleic acids is forbidden. The reason is that the ribose cannot change from C3'-endo- into C2'-endo-conformation because of the contacts between the hydroxyl group at C2' and other atoms (primarily oxygen in the phosphodiester bond) being too close.
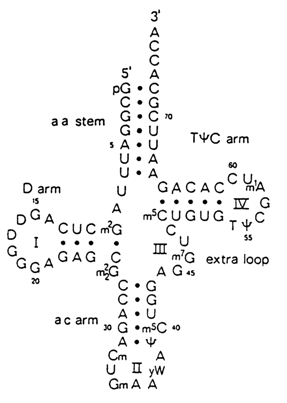
Fig. 8-26. Clover-leaf model of the secondary structure of yeast phenylalanine tRNA.
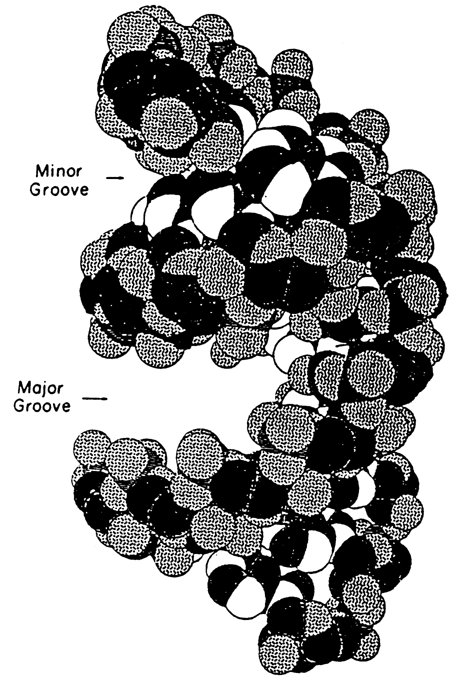
Fig. 8-27. Model of the RNA double helix structure (A-form) (adapted from M. Chastain and I. Tinoco, Jr. Progr. Nucl. Acid Res. Mol. Biol., 41, 131-177 (1991)).
The distance between phosphate groups in base-paired regions of RNA is 0.59 nm (5.9 Å). The grooves of the double helix in RNA differ considerably from those of B-DNA: its major groove is deep and narrow, while the minor groove is shallow and wide. As a result, in contrast to DNA, the main site of interaction with ligands in double-stranded RNA is its minor groove.
If the A
® B transition for double helices of RNA is forbidden, the A®Z like transition is possible; it has been demonstrated that polyribonucleotide duplexes with a repeating GC sequence are in the form of a left-handed helix at a high concentration of salts. Just as in B-DNA and in contrast to A-DNA, a certain degree of microheterogeneity can be discerned in base-paired regions of RNA, which is dependent on the sequence of nucleotides. It manifests itself, for example, in the fact that the anticodon stem of tRNA (Fig. 8-26 and Section 8.2.3) is in the classical A form (11 base pairs per turn), whereas the stem of the D arm contains only about 10 base pairs per turn.Hairpin Loops. Hairpin loops are elements invariably present in the secondary structure of any single-stranded RNA. In the RNAs whose three-dimensional structure has been studied in greater detail, their length varies from three to 15-17 base pairs. There is every reason to assert that the stability of hairpin loops is determining by stacking interactions and hydrogen bonding within the loops. However, there are very few cases where the secondary structure of hairpin loops has been definitively established. At the same time, it is extremely important because. on the one hand, many hairpin loops are involved in tertiary interactions and, on the other, they are the principal components of functional sites in most single-stranded RNAs.
Analysis of the thermodynamic stability of many synthetic hairpins has shown that the most stable structures are those with loops of four or five nucleotides. If a loop consists of only three nucleotides, their ribose moieties
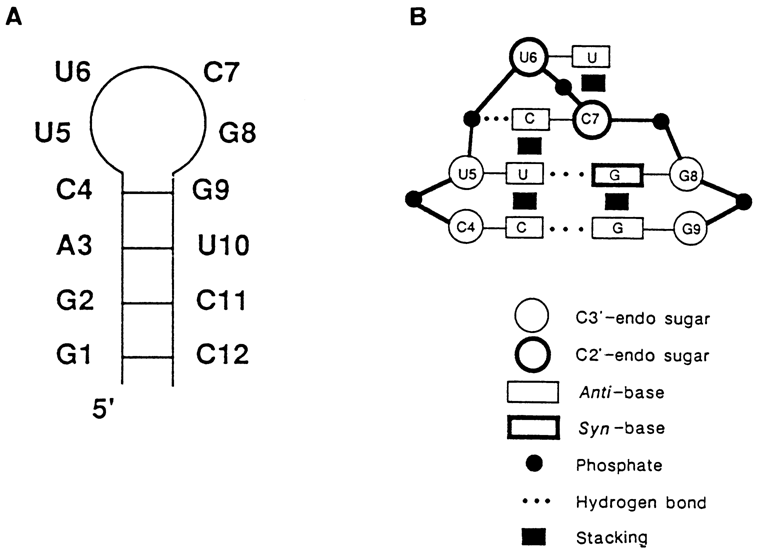
Fig. 8-28. Unusually stable RNA hairpin; (A) secondary structure of the hairpin; (B) schematic diagram of base-pair interactions in its loop (reproduced with permission from C. Cheong, G. Varani, and I. Tinoco, Jr., Nature, 346, 680-682 (1990)).
must acquire the C2'-endo-conformation, which allows individual nucleotides to be spaced as widely apart as possible. Interestingly, the stability of a hairpin as a whole is almost independent of the nucleotide composition of its loop if the conformation of the latter is ensured only by ordinary stacking. However, if more complex intramolecular interactions occur in the loop, its nucleotide sequence may affect structural stability as a whole in a most dramatic fashion.
A good case in point is the hairpin shown in Figure 8-28. It occurs in quite a few RNAs and is marked by high stability. At an ionic strength of about 0.1, its melting point (Tm) is 80.30 C. If a C is replaced by a U in the loop, Tm, will drop by 50 C at once. Investigation of the three-dimensional structure of this hairpin by high-resolution NMR has shown that it has a rather unusual secondary structure which is illustrated in Figure 8-28. Since G8 acquires syn-conformation, it becomes capable of forming a G . U pair with U5 and being heavily involved in stacking with G9. Nucleotides U6 and C7 are in the C2'-endo-conformation with the result that the loop becomes more elongated and C7 can enter into stacking with U5 and form a hydrogen bond with the phosphate group. The latter possibility deserves special attention because we have not yet discussed here any example of formation of specific hydrogen bonds between bases and phosphates, and this, in combination with stacking, may be of great significance for compact packing of loops.
Particular attention should also be given to the anticodon loop of tRNA. It consists of seven nucleotides, and its most distinctive feature is the sharp turn the chain takes between the second and third nucleotides from the 5' end. The other five nucleotides form strong stacking contacts with one another, and the overall conformation of this portion of the loop seems to continue the helical structure of the anticodon stem (Fig. 8-29). The overall conformation of the loop, including the bend, is also stabilized by the intra-loop hydrogen bonds whose formation involves the 2'-OH groups of ribose.
Bulges. Similarly to loops, the bulges forming over double-stranded segments of RNA have been attracting attention as potentially functional sites. This is true, primarily, for single-base bulges containing adenosines whose participation in the RNA-protein interaction was proved on several occasions and which are present in the secondary structure of many RNAs. As was already pointed out in Chapter 7, the extra single nucleotide either protrudes from the double helix or intercalate in it, being involved in stacking with the paired bases. This leads to a certain degree of destabilization of the double helix, which is dependent on the nature of the bulge nucleotide and the composition of the neighboring base pairs. It has been demonstrated that the destabilizing action in the case of bulge purine is more significant than in the case of bulge pyrimidine.
Very little is known about the conformation of larger bulges. There is reason to believe that they destabilize double-stranded segments of RNA to a greater degree than single-base bulges. As regards much larger bulges, their conformation is likely to be stabilized by intra-bulge interactions typical of hairpin loops.
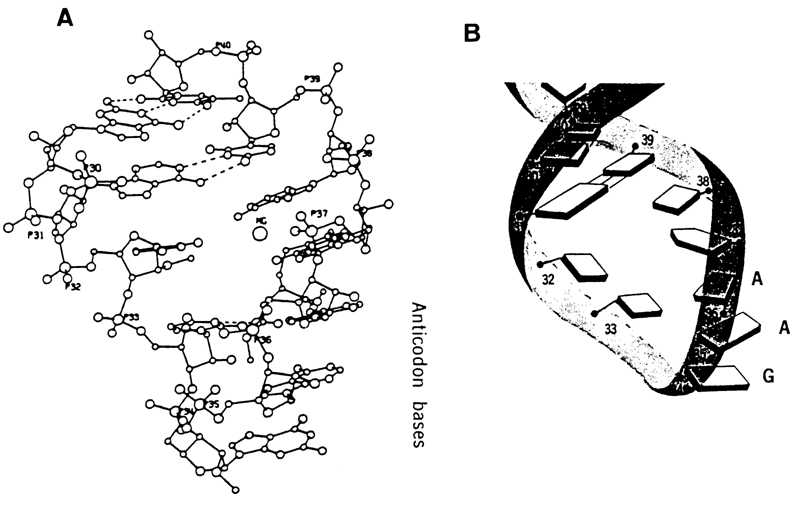
Fig. 8-29. (A) 3D structure and (B) schematic diagram of three-dimensional structure of the anticodon loop of yeast phenyl-alanine tRNA (adapted from S.-H. Kim and J. L. Sussman, Nature, 260, 645-646 (1976)).
Internal Loops. These motifs arise whenever bases that are not complementary in the Watson-Crick sense find themselves opposite each other in a double-stranded segment of RNA. Therefore, the stability of an internal loop will depend on whether the bases in the opposite strands become paired as well as on the degree to which such pairing disturbs the geometry of the helix. The least disturbance in the double helix is caused by formation of a G. U pair. It is thermodynamically more advantageous than formation of any other noncanonical pair. What is more. X-ray structural analysis of short oligonucleotide duplexes with G. U pairs has shown that they do not cause any perceptible disturbances in the geometry and stacking of other pairs in the duplex. This is precisely why, if G and U are in complementary strands of RNA opposite each other, they are represented in a base-paired form. It should be remembered, however, that incorporation of a G . U pair always destabilizes the double helix of RNA and its replacement by an A . U pair raises the Tm of this RNA region by several degrees.
It has been speculated that other bases may also be paired in internal loops, for example, A with G or another A, a protonated A with C, or U with another U. The formation of such pairs is usually postulated proceeding from the assumption that their bases do not lend themselves to modification with chemical agents. However, the screening of bases may also stem from the fact that a particular internal loop is involved in tertiary interactions.
Junctions. If we take portions of the RNA secondary structure where branches are linked together, the most interesting and not yet discussed type of interaction is coaxial stacking of double-helical segments. It occurs by virtue of the fact that the base pairs at the ends of the double-stranded segments are in contact with each other, become involved in stacking, and one helix thus becomes a continuation of the other. The most vivid and yet rigorously proved example of such junction is provided by paired coaxial interactions between helical portions of tRNA. Similar junctions are believed to exist in many other RNAs as well.
8.2.3 Macromolecular Structure of Transfer RNAs
On several occasions we already mentioned the "clover-leaf" model and its constituent secondary structure elements. It should be noted, however, that after the nucleotide sequence of the first tRNAs had been established, this model ceased to be as obvious as it was purported to be and serious efforts had to be made to corroborate it. Indeed, analysis of the nucleotide sequences of tRNA available at that time had led to three hypothetical models of secondary structure: (a) a long hairpin with defects; (b) several (two or three) separate short hairpins; and (c) several (three or four) short hairpins forming a "clover leaf" (Fig. 8-30).
These three hypothetical models were subjected to thorough verification by various chemical and physical methods. It has been found that the "cloverleaf" model is most consistent with the experimental results.
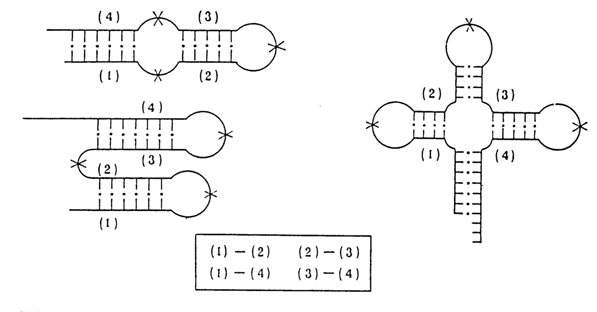
Fig. 8-30. Three possible ways of organization of the secondary structure of tRNA and proof of validity of the clover-leaf model.
Firstly, the "clover-leaf" model fits well the physico-chemical data on the degree of coiling (number of paired bases) and the length of helical segments in different total and individual tRNAs.
Secondly, as indicated by theoretical calculations, the "clover-leaf" model for most tRNAs with a known primary structure is a rather advantageous type of secondary structure in thermodynamic terms (i. e., corresponds to the minimum of free energy).
Thirdly, strong arguments in favor of this model were supplied by analysis of oligo- and polynucleotide fragments resulting from hydrolysis of tRNAs with specific RNases. It is known that most RNases hydrolyse primarily the phosphodiester bonds in single-stranded segments of RNA. Thus, partial enzymatic digestion of tRNAs makes it possible to obtain large fragments retaining their capacity for reassociation and restoration of the double-stranded segments that existed in the starting tRNA molecule. Figure 8-30 shows schematically that the pattern in which quarters of tRNA can be reassociated is consistent with only one of the three possible models of secondary tRNA structure, namely, the "clover-leaf" model.
It should be remembered that in accordance with the functions performed by particular tRNA regions or their exact nucleotides distinction is made, in the tRNA molecule, between single-stranded anticodon loop, dihydrouridyl loop (D-loop), extra loop, T
yC-loop, and acceptor end (see Figure 8.26).Numerous experiments have shown that most bases in single-stranded portions of tRNA, inferred from a "clover-leaf"-type model, are not amenable to chemical modification. A case in point is bases in the T
yC-loop.The different accessibility of nucleotide sequences in single-stranded segments of tRNA can also be demonstrated by the method of complementary oligonucleotide binding, which is quite popular in studying the macromolecular structure of nucleic acids. The method involves the following procedure.
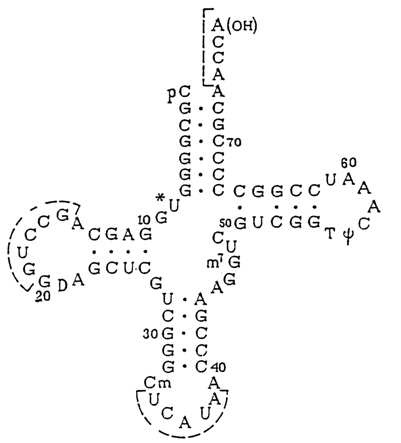
Fig. 8-31. Probing of three-dimensional structure of E. coli tRNAMet by association with complementary oligonucleotides. Sites available for binding of oligonucleotides are shown by broken lines.
Having a set of short oligonucleotides (most commonly, penta- and hexanucleotides) complementary with respect to the single-stranded segments predicted by the secondary structure of a particular RNA, one can measure the association constant (Kass) of these nucleotides with the RNA. The measurements are conducted under conditions optimal for formation of double-stranded complexes. Surface portions of the macromolecule will be characterized by maximum value of Kass for oligonucleotides. In this case, the nucleotide composition of the probed segments is taken into account, of course, in view of the fact that the presence of G . C pairs in the emerging oligonucleotide-RNA complex may bring Ka,, up by several orders of magnitude. The results produced by thus method agree well with those of chemical modification of tRNA. Moreover, they provide information on accessibility not only of individual nucleotides but also some RNA segments (Fig. 8-31).
Studies of tRNA macromolecules by such methods have led the investigators to a firm conclusion that the separate loops of the "clover-leaf" form a specific tertiary structure common to all tRNAs.
In 1973-75, the tertiary structure of tRNA was solved in brilliant experiments conducted by A. Rich's and A. Klug's groups who investigated, by X-ray structural analysis [with a resolution of up to 0.25 nm (2.5 Å)], two crystalline forms of yeast phenylalanine tRNA (tRNAPhe) and arrived at virtually identical tertiary structure models for this tRNA (Figs. 8-32 and 8-33).
According to the Rich-Klug model, the macromolecule of tRNA has a simplified L shape, its major functional sites, anticodon loop and acceptor end, terminating the L at both limbs. The distance between the two is 7.6 nm (76 Å). The double-stranded segments adjacent to the acceptor end and T
yC-loop form a single double helix. This helix forms almost a right angle with the double-stranded segment of the D-loop and the following double-stranded part of the anti-codon "hairpin". The two main linear portions of the macromolecule are held together by complex intertwining of the D- and TyC-loops. All double-stranded segments of the molecule belong to the A form (i. e., their riboses are in 3'-endo-conformation). At the same time, some nucleotides have been found to contain a ribose moiety in 2'-endo-conformation in single-stranded segments.By far the most important result of X-ray structural analysis of tRNAs seems to be evaluation of the specific internucleotide interactions responsible for the tertiary structure of their macromolecules. The main types of intermolecular interactions have turned out to be the same as in formation of the classical secondary structure of nucleic acids - that is, stacking and complementary interaction of heterocyclic bases. However, base pairing and the associated hydrogen bonding in this case involve donor and acceptor groups other than those participating in Watson-Crick pairing. Consider a few examples.
Figures 8-32 and 8-33 illustrate nucleotides occupying different parts of the "clover-leaf" but coming close together and interacting during formation of the tertiary structure of tRNAPhe. For instance, pseudouridylic acid
y(55) (from the TyC-loop) forms an inverted Hoogsteen pair with G(18) from the D-loop, whereas the 1-methyladenine m1A(58) is paired with the thymine T(54) (Fig. 8-34).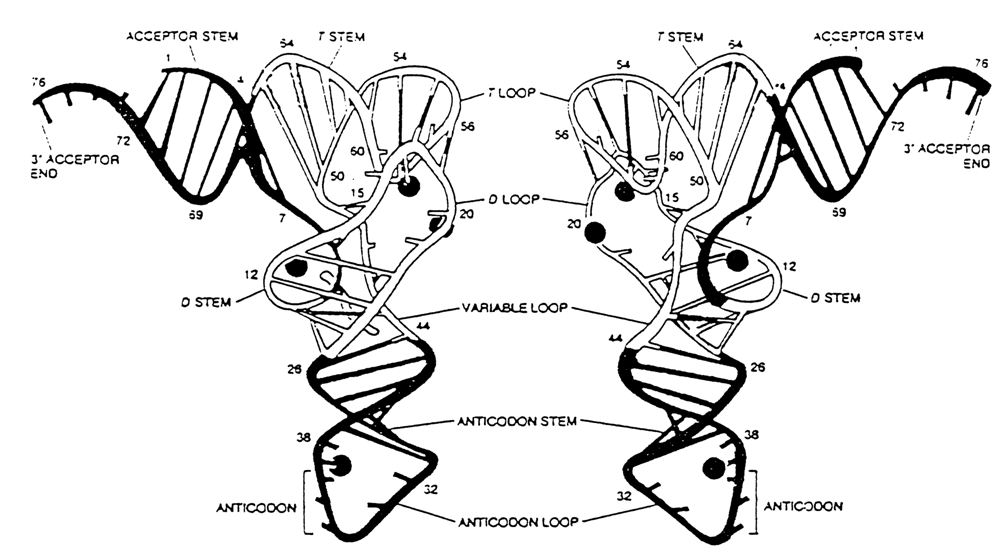
Fig. 8-32. Tertiary structure model proposed by Rich and coworkers for tRNAPhe. The sugar-phosphate backbone of the molecule takes the form of a ribbon. The macromolecule is shown from two opposite sides. Hydrogen bonds are shown as junctions between the strands. The molecular fragments and bonds involved in the tertiary structure are shown in black.
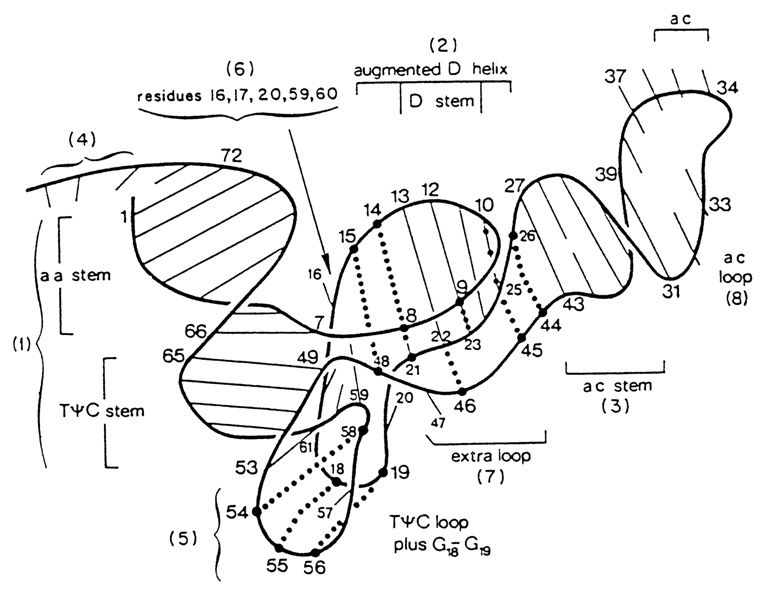
Fig. 8-33. Tertiary structure model proposed by Klug and coworkers for tRNAPhe. The sugar-phosphate backbone is shown as a thick line; the long thin lines stand for hydrogen bonds; the dotted lines stand for the hydrogen bonds involved in the tertiary structure; the short thin lines indicate the position of bases not linked by hydrogen bonds.
G(57) finds itself between the G(I8)-
y(55) and Watson-Crick pairs; G(19) (from the D-loop) and C(56) (from the TyC-loop) become involved in pronounced stacking with two neighboring guanines G(18) and G(19) (Fig. 8-35).Another interesting contact arises at the site where the double-helical segment adjacent to the D-loop approaches the extra loop. Here, the minor base M7G(46) forms a pair, across the broad groove of the helix, with G(21) which, in turn, is paired with G(11) (Fig. 8-34). In this case, the positive charge at m7G(46) is neutralized by the phosphate group at A(9).
A number of unusual base pairs emerge in that part of the tRNA molecule where its chain forms a deep fold (between the 7th and 10th b.p.). This part features, for example, an inverted Hoogsteen pair U(8)-A(14) and an unusual pair between C(48) and G(15) (Fig. 8-34).
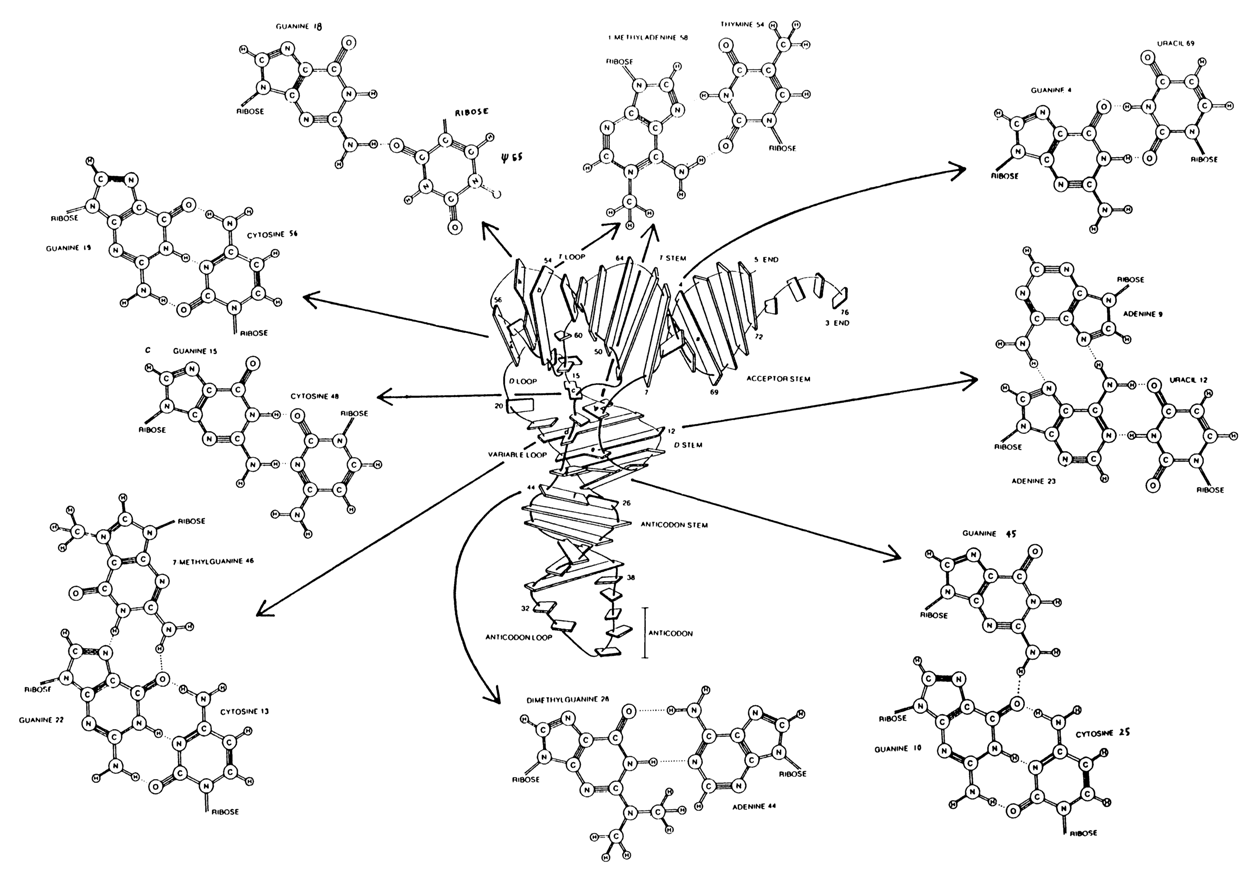
Fig.8.34. Tertiary base pairs in the crystal structure of yeast tRNAPhe (reproduced with permission from S.-H. Kim in: S. Neidle, ed., Topics in Nucleic Acid Structure, part I, pp.83-112, MacMillan 1981).
In addition, certain parts of the molecule become sites of hydrogen bonding between neighboring riboses as well as binding of neighboring phosphate groups by ions of bivalent metals.
As already mentioned, significant progress has been made over the past few years in studying tRNA by NMR spectroscopy. This technique makes it possible to resolve signals from a large number of protons, including those involved in hydrogen bonding and belonging to NH groups practically in all base pairs emerging during formation of the secondary and tertiary tRNA structures. NMR spectra indicate that tRNA molecules in solution and in the crystalline form have closely similar, if not identical, structures. Thus, they allow tRNA samples that cannot be crystallized to be investigated with a nearly atomic resolution. Moreover, analysis of NMR spectra permits one to separately observe almost every base pair in a tRNA molecule, which is extremely important in studying the mechanism of its functioning.
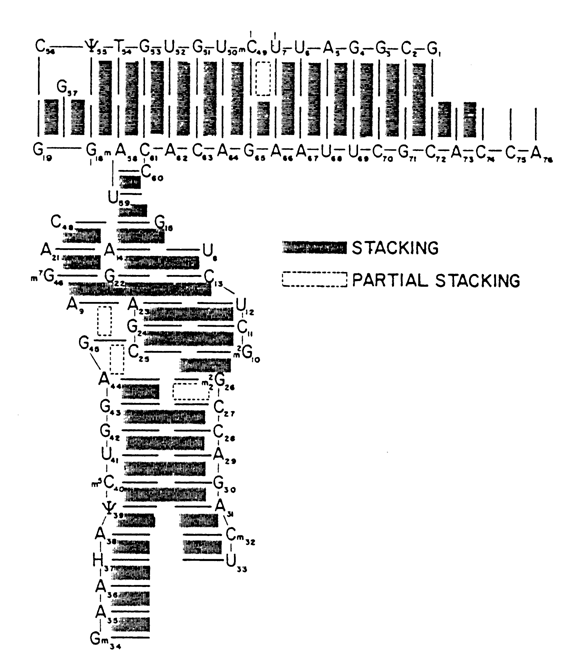
Fig. 8-35. Stacking interactions in tRNAPhe. The T
yC-loop occupies the upper left corner. The thin lines interconnect adjacent bases in the polynucleotide chain. The thick lines indicate paired bases; bases not involved in stacking are not shown. It can be seen how helical portions associated with the acceptor end and TyC-loop are interlinked as well as how the D- and TyC-loops are intertwined (reproduced with permission from S.-H. Kim, Progr. Nucl. Acid Res. Mol. Biol., 17, 181-216 (1976)).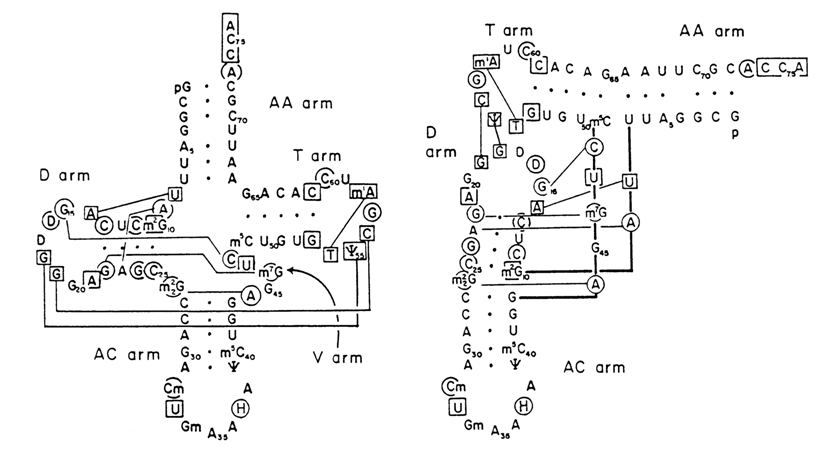
Fig. 8-36. Scheme showing the conservation of tertiary interactions in tRNA. The solid lines interconnect bases paired in the tertiary structure of tRNA. The boxed bases occupy permanent sites in all tRNAs, while the encircled ones occupy those sites in the polynucleotide chain where all tRNAs contain only purines or only pyrimidines.
Importantly, many bases involved in the formation of the tertiary structure of tRNAPhe are universal in the sense that they occur at the same positions of the polynucleotide chain in all tRNAs that have been studied (Fig. 8-36). Hence, all tRNAs are characterized by a macromolecular structure consistent with the Rich-Klug model. This has been convincingly supported by the results of X-ray structural analysis of other crystalline tRNA structures as well as those in solution. Minor dissimilarities have been revealed between different tRNAs, primarily near the anticodon loop and acceptor end of the molecule. It has also been found that the angle made by two axes of the limbs of the L-shaped tRNAAsp structure is apparently 100 greater than that of tRNAPhe (Fig. 8-37).
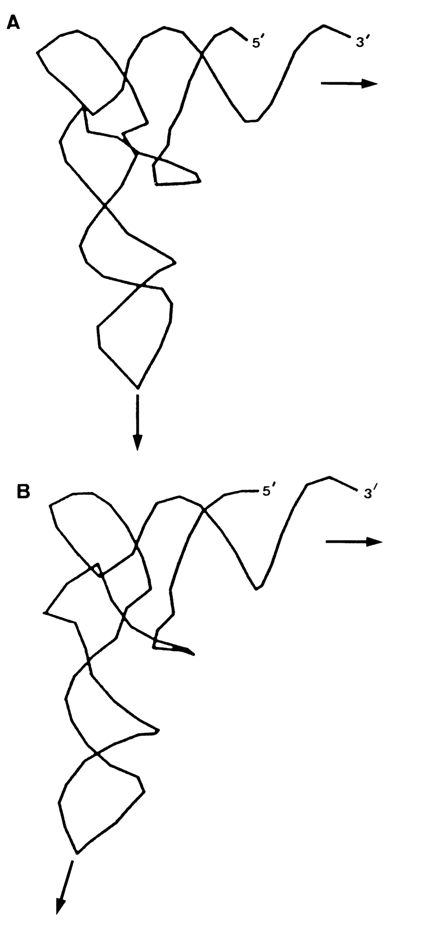
Fig. 8-37. Comparison of backbone conformation of (A) tRNAPhe and (B) tRNAAsp (adapted from D. Moras, Curr Opinion in Struc. Biol., 1, 410-415 (1991)).
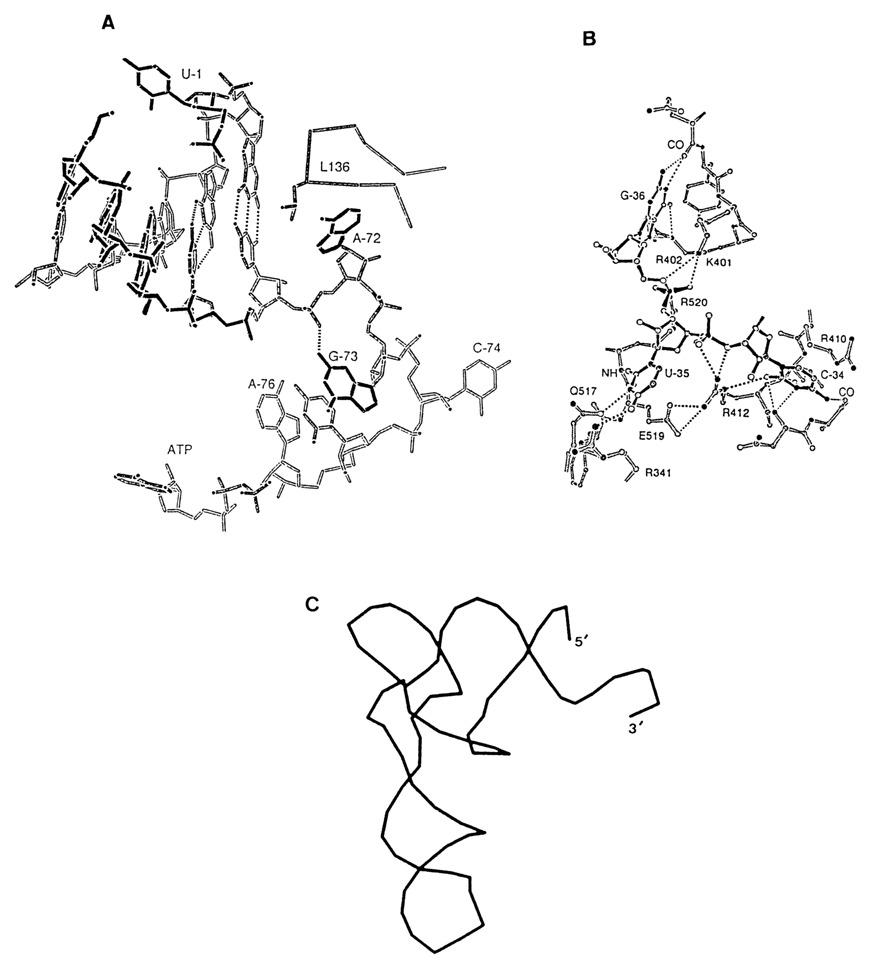
Fig. 8-38. (A) Structure of the acceptor strand of the E. coli tRNAGln in the complex with glutaminyl-tRNA synthetase (adapted from M. A. Rould, Science, 246, 1135-1142, 1989); (B) structure of an anticodon loop of E. coli tRNAGIn in the complex with glutaminyl-tRNA synthetase (adapted from M. A. Rould, Nature, 352, 213-218, 1991); (C) schematic representation of the tRNAGln backbone in the complex with glutaminyl-tRNA synthetase (adapted from D. Moras, Curr. Opinion in Struc. Biol., 1, 410-415, 1991).
It has often been suggested that the tertiary structure of a functioning tRNA undergoes drastic changes. Indeed, when a complex of tRNAGln with glutaminyl-tRNA synthetase is formed, the last base pair in its acceptor stem breaks down and the acceptor end forms a loop directed toward the anticodon (Fig. 8-38). The bases in the anticodon loop, forming part of the anticodon, pass into a more open conformation, which ensures a more intimate contact with the protein. What is not known, however, is how common such conformational changes are. For example, in a complex with specific aminoacyl-tRNA-synthetase. the tertiary structure of tRNAAsp is virtually indistinguishable from that of free tRNA.
8.2.4 Three-Dimensional Structure of High-Molecular Weight RNAs
Transfer RNAs are the only representatives of this class of nucleic acids that have been successfully crystallized and studied by X-ray structural analysis with atomic resolution. In the case of RNAs comparable in size with tRNA, such as 5S ribosomal RNAs or small nuclear RNAs, high-resolution NMR was applied only to study the conformation of rather short fragments (the separate elements) of their molecules.
However, as we go to larger RNAs, rather serious difficulties arise and in order to establish their three-dimensional structure we have to use many indirect mutually complementary methods.
As a rule, the investigator begins with establishing the nucleotide sequence of the RNA. The next step is a computer search for possible models of secondary structure of the same RNA. Such search involves well proven algorithms for constructing various possible elements of the RNA secondary structure, considered in 8.2.2, and evaluating their thermodynamic stability. They are based on a wealth of experimental data obtained on synthetic oligoribonucleotides and make it possible to estimate the length of double-stranded segments, the sequence of G.C, A.U and G.U pairs in the latter, the size of the bulges, and so on.
Unfortunately, calculations of this kind usually result in numerous secondary structure models with closely similar values of free energy for a given RNA (the number of such models being extremely high for high-molecular weight RNAs). However, the theoretical search for the model may be continued if the nucleotide sequences for at least one more (better several) RNA of the same type but isolated from a different source are known. In such a case, one can successfully resort to the so-called "phylogenetic" approach which is based on the assumption that the RNAs performing the same function in the cell must have identical secondary and tertiary structures. Then, one must select models of the same type among those calculated for two (or more) RNAs.
The applicability of the "phylogenetic" approach was for the first time demonstrated while studying the structure of tRNA. It was found that the secondary structure of any tRNAs, no matter how their nucleotide sequences vary, is consistent with the universal "clover-leaf" model. Even more impressive are the results of using this approach to ribosomal RNAs, when a single model had been selected for RNA of each type among hundreds of theoretically possible ones. The exact way in which such selection is carried out at the level of individual elements of the RNA secondary structure is shown in Figure 8-39, while Figure 8-40 illustrates the similarity of secondary structures of the RNA component of ribonuclease P.
The theoretical model of the RNA secondary structure must be experimentally verified. Indeed, one can select a chemical reagent for each heterocyclic base, which interacts selectively only with the atoms or groups involved in hydrogen bonding during complementary pairing of nucleotides. For instance, ketoxal reacts selectively with N1 and the 2-NH2 group of guanine, dimethyl sulfate does so (under certain conditions) with N1 of adenine and N3 of Cytosine, and carbodiimide reacts selectively with N3 of uracil. Consequently, if a particular nucleotide in RNA is modified by one of these reagents, it belongs to the single-stranded segment of RNA. Instances where no modification takes place are harder to interpret - the nucleotide may form part of the double helix but be screened by tertiary interactions as well.
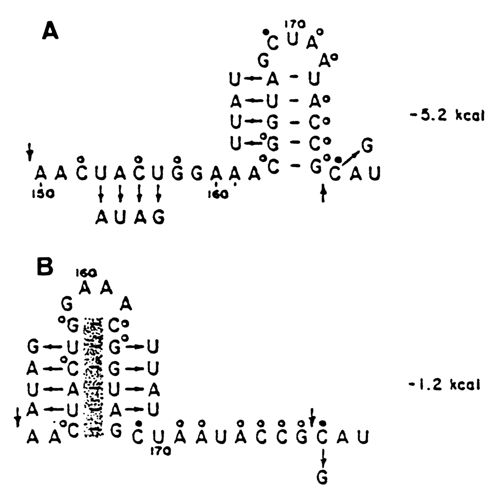
Fig. 8-39. Comparative (phylogenetic) analysis used to select the most probable secondary structure of RNA. Two possible secondary structures of a portion of 16S ribosomal RNA of E. coli (numbering from the 5' end of RNA) are shown. In spite of the fact that the energy of formation of structure (A) is lower, preference should be given to structure (B). Indeed, substitutions in the nucleotide sequence of a similar portion of the molecule of 16S ribosomal RNA of Bac.brevis (shown by arrows) rule out the existence of structure (A). At the same time, in the case of structure (B), the double-stranded portion remains in both RNAs in spite of the nucleotide substitutions. Such substitutions are referred to as compensatory (adapted from R. R. Gutell et al., Progr. Nucl. Acid Res. Mol. Biol., 32, 156-217 (1985)).
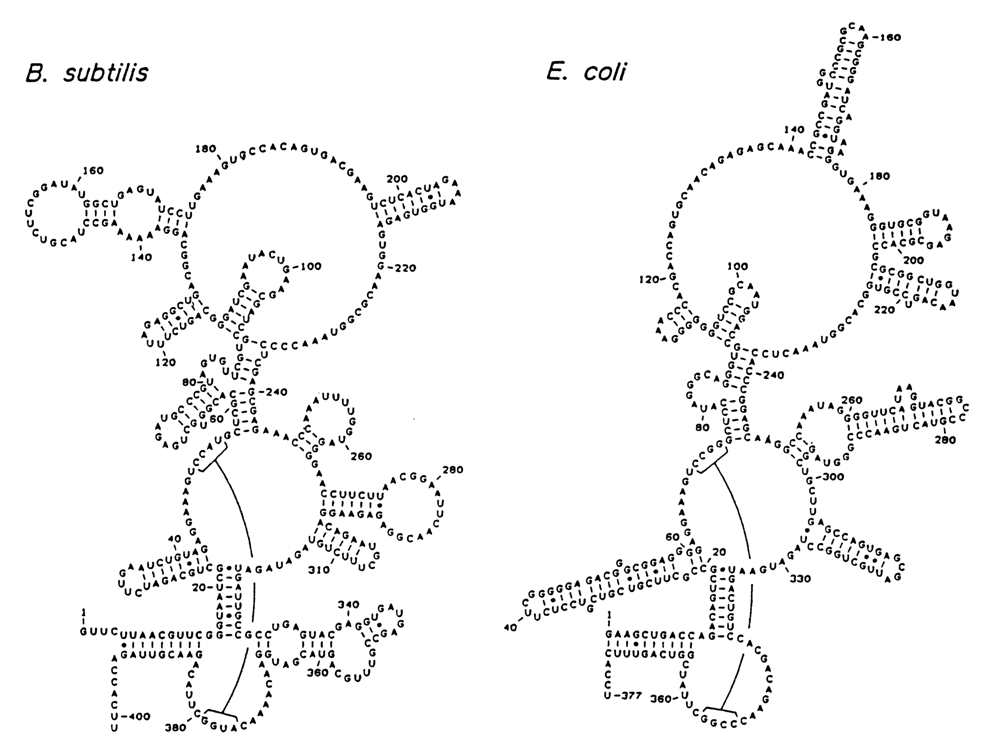
Fig.8-40. Secondary structure of RNA components of E. coli and B. megaterium RNase, proposed on the basis of phylogenetic comparison (reproduced with permission from B.D. James et al., Cell, 52, 19-26 (1988)).
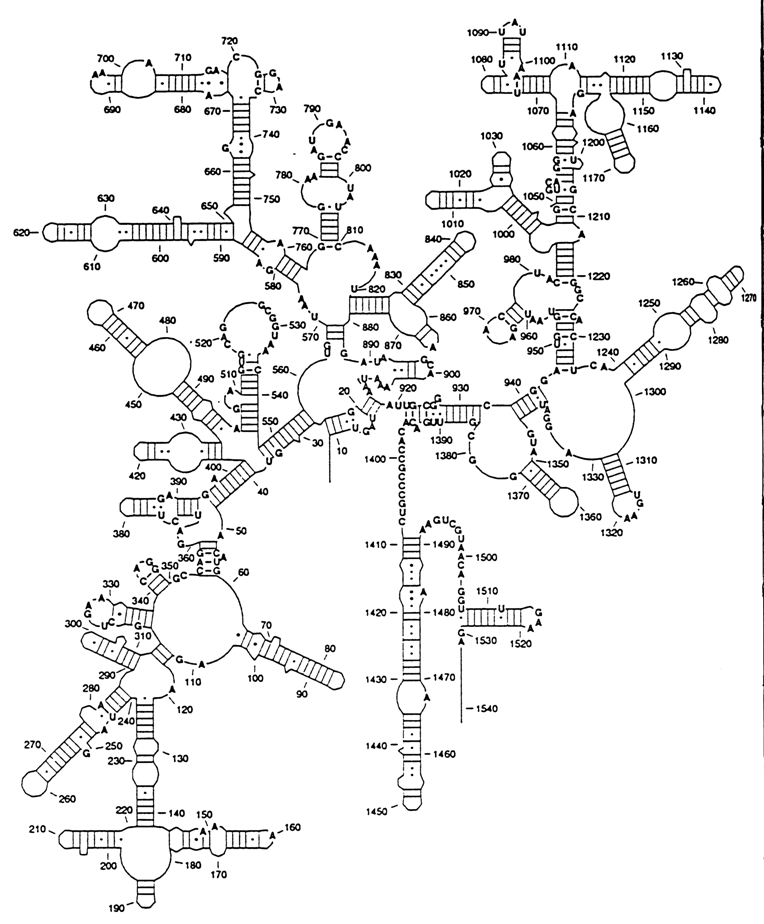
Fig. 8-41. Three-dimensional structure of 5S ribosomal RNA, based on chemical and enzymatic probing (courtesy of E. Westhof).
Similar principles underlie the enzymatic methods of RNA secondary structure analysis. A large number of RNases are known which provide directed hydrolysis of the phosphodiester bonds in single-stranded segments of RNA. Several nucleases have also been discovered which exhibit pronounced affinity toward double-stranded RNAs. Analysis of RNA hydrolysis by these enzymes will reveal the type of secondary structure to which particular nucleotides belong. The results of chemical and enzymatic methods complement each other rather well. Fig. 8-41 represents a 3D structural model of the E. coli 5S ribosomal RNA, derived by combination of the methods described above.
These approaches have been used to derive secondary structure models for many viral and cell RNAs. In spite of the fact that the length of such RNAs equals hundreds of nucleotides, reliable models of their secondary structure have been produced in full agreement with experimental data. The new property brought to light by secondary structure analysis of such large RNAs is the subdivision of their macromolecules into structural domains or, in other words, large molecular fragments having a more or less independent three-dimensional structure. Figure 8-42 shows the universal structure of an RNA from a small ribosomal subunit.
Trying to establish the tertiary structure of high-molecular weight RNAs is an even more challenging task. For want of a better approach, one must proceed from the principles on which the tertiary structure of tRNAs is based. In other words, the following assumptions are to be made as regards any single-stranded RNA:
1. Elements of the RNA secondary structure are mutually arranged in such a fashion so as to ensure a maximum degree of base stacking in the macromolecule as a whole.
2. Contacts between individual secondary structure elements are based on several types of so-called "tertiary" intramolecular interactions: (a) those involving formation of additional, often non-Watson-Crick base pairs between nucleotides in single-stranded segments spaced widely apart (in the primary and secondary structures) and base triplets between nucleotides in single- and double-stranded segments; in the latter case. participating in hydrogen bonding are the N7 atoms of purine bases as well as the functional groups at their C6 atoms, already taking part in formation of a single hydrogen bond in the Watson-Crick pair; (b) those involving additional ("tertiary") stacking interactions after intercalation of bases from one segment between two neighboring bases of another single-stranded segment; and (c) those involving formation of additional hydrogen bonds between the 2'-OH groups of ribose and bases as well as other groups of the sugar-phosphate backbone.
3. The tertiary structure of RNA is stabilized by bivalent metal ions which become bound not only with phosphate groups but also with bases.
Fig. 8-42. Universal model of RNA secondary structure from a small ribosomal subunit (reproduced with permission from M. Noller, in The RNA World, R. Gesteland and J. Atkins, eds., pp. 137-156, Cold Spring Harbor Lab. Press, 1993).
These principles are not sufficient, of course, to deduce the three-dimensional structure of an RNA from its primary and secondary structures alone. As will be demonstrated in the following chapter, chemical modification data are instrumental, too. Valuable information is also provided when bifunctional alkylating agents, such as bis(2-chloroethyl)methylamine or "nitrogen mustard", are used. They cross-link bases spatially arranged next to each other but belonging to different segments of the RNA chain. Then the RNA is fragmented and the cross-linked oligonucleotides are isolated and sequenced.
There is no doubt that the "phylogenetic" principle must also hold for the tertiary RNA structure. What is more, it can be assumed that two or more RNA molecules differing in secondary structure but performing similar functions in the cell will have similar tertiary structures. A case in point is RNAs of plant viruses. Some of them, such as turnip yellow mosaic virus (TYMV) RNA, are aminoacylated at the 3' end by aminoacyl-tRNA-synthetases, similarly to tRNA. The secondary structures of the 3'-terminal segment of TYMV RNA and tRNA are similar and yet display significant differences. As can be seen from Figure 8-43, these differences disappear almost completely when this segment of the viral RNA is integrated into the tertiary structure. Here we encounter yet another essential element of macromolecular RNA structure, known as the pseudoknot. Pseudoknots result from complementary pairing of hairpin-loop bases with those in the single-stranded segment outside the hairpin structure (Fig. 8-44). The possibility of such pairing has been demonstrated on model oligoribonucleotides. Pseudoknots represent a widely occurring and highly important element of three-dimensional structure of many RNAs.
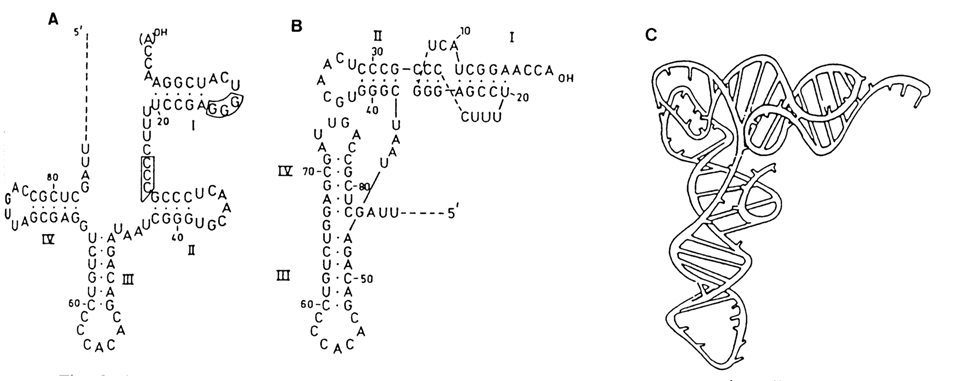
Fig. 8-43. Secondary and tertiary structure of the 3'-terminal segment of turnip yellow mosaic virus (TYMV) RNA. A - Clover-leaf structure; the regions capable of forming pseudoknots are boxed; B - L-shaped structure; C - three-dimensional model (adapted from C. Pleij, TIBS, 15, 143-147, 1990).
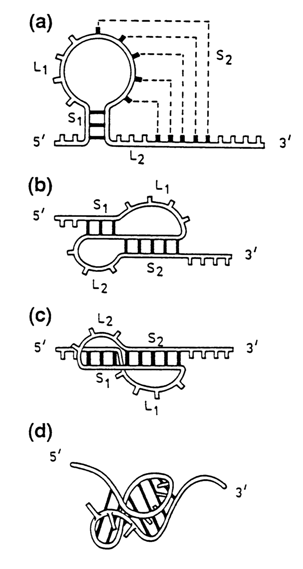
Fig. 8-44. Formation of a pseudoknot (a) - starting structure; the broken line indicates complementary bases in the potential pseudoknot; (b) and (c) - formation of the S2 stem. Note the coaxial stacking of S1 and S2 in (c); (d) - three-dimensional structure of the pseudoknot (reproduced with permission from C. Pleij, TIBS, 15, 143-147 (1990)).