![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
7.4 Structure of Double- and Multiple-Stranded Polynucleotide Complexes
Single-stranded polynucleotides with complementary sequences may form helical complexes under appropriate conditions.
The simplest and most typical example of such structures is be provided by complexes formed by complementary homopolymers - that is, poly(A) and poly(U) or poly(G) and poly(C). The composition of a complex depends on the ionic strength of the solution in which it is formed. For instance, at a low ionic strength in the absence of magnesium ions, a double-stranded complex of poly(A) and poly(U) [i.e., a poly/A).poly(U) complex] can be observed. In a 0.01 M magnesium chloride solution they form a complex made up of one poly(A) strand and two poly(U) ones [i.e., a poly(A). 2poly(U) complex].
The complex formation process is always accompanied by a hypochromic effect. This phenomenon is put to practical use in determining the composition of complexes. By using this approach it was demonstrated that transitions of the types
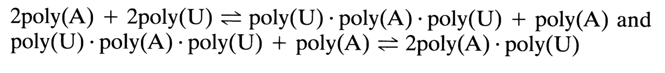
take place between different complexes.
Recently, a large number of double- and triple-stranded polynucleotide complexes have been investigated by X-ray structural analysis. The results of these investigations and some other examined properties to be discussed in what follows indicate that these compounds have the shape of regular double or triple helices. The stability of helical structures is determined by the stacking of adjacent bases and complementary interactions between bases facing each other in different strands.
In the case of double-stranded complexes, the second type of interaction is associated with formation of classical (or, as they are sometimes also referred to, canonical) Watson-Crick pairs (see 7.2). Polynucleotide strands in such complexes are antiparallel, which is to say that if we move along one of the strands in the direction of the phosphodiester 3'
® 5' bonds, in the opposite strand our movement will be in the direction of the phosphodiester 5' ® 3' bonds. In triple-stranded complexes, the pairing of bases involves the N7 atom of the purine ring. An example of such a base-paired poly(A). 2poly(U) complex is shown in Figure 7-21.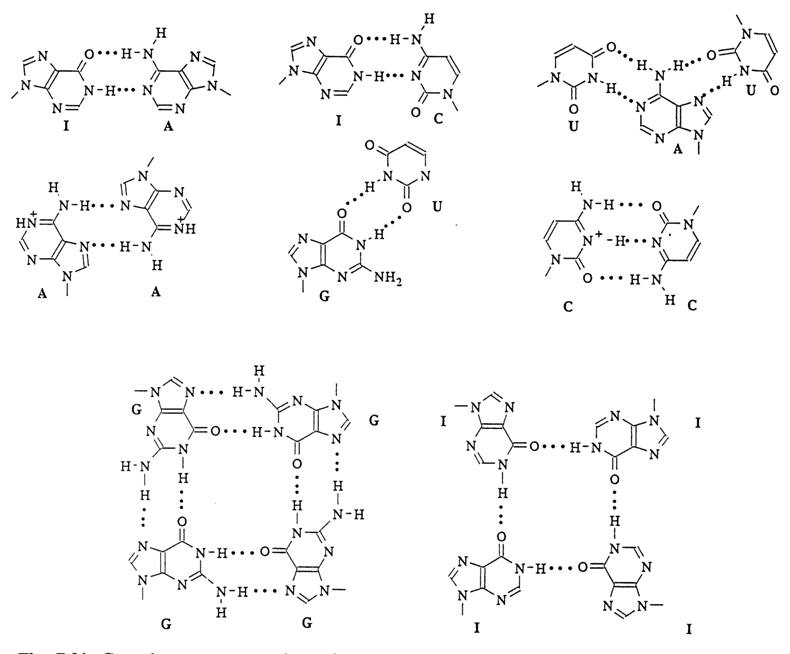
Fig. 7-21. Complementary complexes formed by bases in different double, triple and quadruple polynucleotide helices.
Interestingly, some homopolymers consisting of nucleotide units with bases that are non-complementary from the standpoint of the Watson-Crick concept are also capable of forming double-stranded complexes between themselves. A case in point is complexes of polyinosinic acid with poly(A) and poly(C) (Fig. 7-21).
What is more, at pH < 6.5 poly(A), poly(C) and poly(I) may form regular helical complexes through self-association. The complexes are additionally stabilized by the salt bonds between the phosphate groups of one strand and protonated bases of the other. In the case of poly(A) and poly(C), for example, the pairs shown in Figure 7-21 emerge. In contrast to the ordinary double-stranded complexes with Watson-Crick base pairs, the polynucleotide strands in complexes formed with the participation of protonated bases are parallel (i.e., the direction of their phosphodiester bonds is the same).
The most complex structure is observed in the highly stable complexes formed by poly(G). They are composed of four parallel strands. The ability of guanine derivatives to form quadruplex-type aggregates shown in Figure 7-21 manifests itself already at the mononucleotide level. In recent years, interest in polynucleotide complexes containing G-quartets has risen substantially because G-rich sequences have been revealed in terminal portions of linear chromosomes, known as telomers. Four-stranded DNA structures stabilized by cyclic hydrogen bonding of guanines are covered at greater length in Chapters 8 and 9.
Polyinosinic acid is also capable of forming four-strand complexes. The latter, however, are less stable than poly(G)4 complexes because in this case the tetramer is stabilized only by four hydrogen bonds (Fig. 7-21).
Of particular interest are double-stranded complexes resulting from G pairing with U (or T). The structure of this pair is also illustrated in Figure 7-21. It occurs in double-stranded portions of RNA (Chapter 8) and forms during codon-anticodon interactions. The stability of such duplexes is low (much lower as compared, for example, to complexes formed by pairing of A with U), polyribonucleotides forming more stable complexes than their deoxy analogs.
At low temperatures (below 150 C), poly(U) strands are capable of folding back upon themselves to form numerous imperfect double-stranded structures (of hairpin type) which are in equilibrium (Fig. 7-22). As the temperature rises, these helical structures transit into a single-stranded form in a cooperative manner (see below).
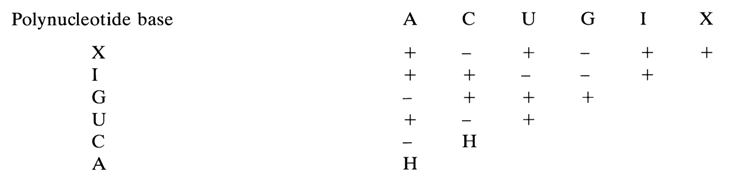
Evidence showing that helical complexes with two and more strands can emerge from various homopolynucleotides is summarized below ("+" indicates that a complex is formed, "-" indicates that no complex is formed, and "H" stands for a complex whose formation involves protonated forms of the base).

Fig. 7-22. Formation of helical structures in poly(U) at low temperature and high Mg2+ concentrations.
An important feature of the intramolecular forces stabilizing double- and triple-stranded helical structures is their cooperative nature. In the case of the simplest complexes formed by complementary homopolynucleotides, this means that under the effect of certain external factors denaturing the complex all base pairs in the helical structure break down simultaneously. Roughly, the term "phase transition" can be applied to this process.
In the case of double-stranded complexes with a more complicated nucleotide composition of complementary chains, the term "cooperativity" is used implying that the helical structure is also denatured within a rather narrow range of external changes.
Let us now consider the process of denaturation of double-stranded polynucleotide complexes in greater detail. Studying this process is extremely important because it provides a wealth of information on the nature of the factors determining the structure of these compounds. The process of denaturation of these complexes is often called helix-coil transition because the tertiary structure of single-stranded polynucleotides is close to a random coil.
The most commonly used method for breaking down double-stranded polynucleotide molecules is their thermal denaturation. Its course is usually monitored by changes in the optical properties of the complexes. Since the breakdown of an ordered helical structure is accompanied by a hyperchromic effect, it is more convenient and simpler to observe the changing optical absorption of these compounds in the UV region.
Since the denaturation of complexes occurs in a narrow temperature interval, the optical absorption-temperature curves are referred to as melting curves. Figure 7-23 represents a melting curve for a poly(A) . 2poly(U) complex.
The temperature at which the percentage of the helical and denatured portions are equal is known as melting temperature and is denoted Tm (on melting curves more complex than the one shown in Figure 7-23 Tm is determined as the temperature at which the increase in optical absorption is 50% of the final level). Another characteristic of denaturation of a helical structure is the width of the melting range DTm. In the case of the poly(A) . poly(U) complex, DTm is small (about 20 C), which is indicative of a high degree of cooperativity of melting of this complex.
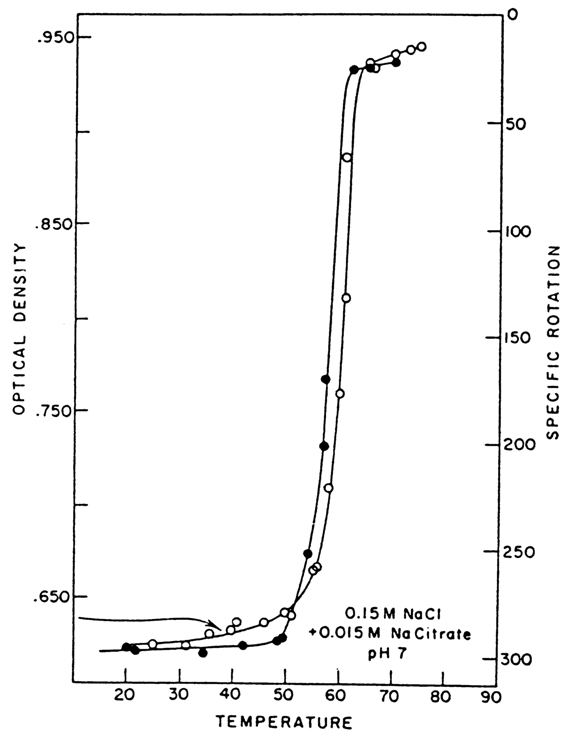
Fig. 7-23. Thermal denaturation profile of poly(A) . 2poly(U) complex at a high ionic strength, determined by UV absorption and ORD at 259 nm (adapted from J. R. Fresco, in: Information Macromolecules, H. J. Vogel, V. Bryson and J. O. Lampen, eds., Acad. Press, N. Y., pp. 121-142, 1963).
Double-stranded polynucleotides with different nucleotide compositions have different Tm. As can be inferred from the above data (see, e.g., 7.2), G . C is a more stable pair than A . U. Besides, stacking interactions which involve cytosines are more pronounced than that involving uracils. Therefore, as the G . C pairs increase in number, Tm of the complex goes up.
It has been established that double-stranded complexes formed by complementary polyribonucleotides most commonly have higher Tm than those formed by the corresponding polydeoxyribonucleotides. We have already mentioned that the stacking of bases in oligoribonucleotides is more pronounced, as compared to oligodeoxyribonucleotides with the same nucleotide sequence.
The stability of double-stranded polynucleotides (as well as their Tm) increases with the polynucleotide chain length. For example, if we take a poly(A) . poly(U) system at room temperature, a double-stranded complex may exist if it is made up of hexa- and longer oligonucleotides. The differences in Tm diminish as the chain becomes longer and become negligible when the chain length reaches approximately 20 nucleotides.
Tm of double-stranded complexes is strongly dependent on the ionic strength of the solution. As a rule, it increases with salt concentration. The reason is that the electrostatic repulsion of negatively charged phosphate groups (located both in one strand and in opposite ones) exerts a strong destabilizing effect on the structure of double- and triple-stranded complexes. Moreover, these complexes may exist only if the phosphate groups are screened by cations. The screening effectiveness depends on the constants of dissociation of the corresponding metal phosphates. This is why most bivalent cations (Mg2+, Ca2+ , Mn2+, etc.) stabilize the structure of complexes much more efficiently than univalent ones. Usually, there is a linear relationship between the melting point of double-stranded polynucleotides and the logarithm of salt concentration in the solution. It should be pointed out that some bivalent cations (e.g., Cu2+ ) lower the melting temperature of natural and synthetic double-stranded polynucleotides.
The stability of double-stranded helical polynucleotide complexes and their Tm are virtually independent on pH of the medium in the range from 5.5 to 9.0. However, at lower or higher pH values the complexes become less stable, which has to do with ionization of the bases.
Tm of double-stranded complexes is materially affected by the presence of organic solvents, urea, guanidine, and some anions (CIO4-, ClO3-. CC13COO-) in high concentrations. All these substances upset the interplanar (stacking) and/or complementary interactions between bases.
While studying the process of thermal denaturation of complementary polynucleotide complexes, one can glean important quantitative characteristics of the intramolecular interactions in them. For instance, the following relation has been obtained for homopolynucleotide complexes:
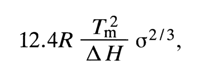
where (
s is the cooperativity factor, R is the universal gas constant, and DH is the enthalpy of the complex's transition from ordered to denatured state.The cooperativity factor
s, in turn, is related quantitatively to the change in the free energy of stacking of two separate hydrogen-bonded base pairs, E, as follows (T is absolute temperature):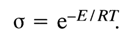
From the known value of
DH and experimental melting curve one can calculate the value of E. For double-stranded homopolynucleotide complexes it is about 29.4 kJ/mol (7 kcal/mol). Knowing the basic factors responsible for denaturation of double-stranded polynucleotide complexes we now can formulate the optimal conditions for formation of such complexes: (1) the temperature must be substantially lower than Tm; (2) a solution with high ionic strength must be used; (3) the concentration of single-stranded polynucleotides must not be lower than 10-5 M.