![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
7.2 Intermolecular Interactions Between Heterocyclic Bases
The specific macromolecular structure of nucleic acids is determined by two types of interactions between heterocyclic bases: firstly, complementary interactions between bases lying in the same plane (complementary interactions), and, secondly, interplanar interactions between bases arranged one above another (vertical or stacking interactions).
As will be shown below, complementary interactions are based on formation of specific hydrogen bonds and the usual intermolecular forces determined by Van der Waals-London interactions. Stacking interactions are determined primarily by the latter.
It would be appropriate at this juncture to remind that the overall energy of Van der Waals-London interactions, EM, is a sum of the energy of dipole interactions, E
mim2, the energy of a dipole-induced dipole type of interaction, Ema and the energy of dispersion or London interactions, EL, arising between a dipole due to fluctuation and a dipole induced by the former: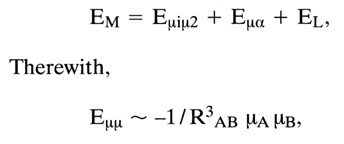
where
mA and mB are dipole moments, RAB is the distance between the interacting dipoles.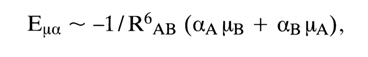
where
aA and aB are polarizabilities.![]()
where 1Aand IB are ionization potentials.
It is extremely important that both complementary and stacking interactions can be studied on very simple systems, such as bases, nucleosides, and nucleotides. It is while studying such systems that the major factors responsible for a particular type of interaction have been identified. Let us now have a closer look at these interactions.
The magnitude of polarizability (usually determined from atomic refractions) is about 0.01-0.012 nm3 (10-12Å3) for uracil, cytosine and thymine and about 0.014 nm3 (14A3) for adenine and guanine. The ionization potentials for all bases lie in range of 1.4-1.6 10-18 J (9-10 eV).
If two interacting bases are considered as two dipoles, the energy of their interaction can be estimated only very roughly. Therefore, the so-called monopole approximation was used to calculate the total energy and the contribution of individual types of interactions to it. The energy of interaction between two bases was regarded as a sum of interactions between atoms of one base and those of the other. Instead of constant dipole moments use was made of net atomic charges, and the total energy is expressed as follows:
![]()
where E
rr is the energy of the Coulombic interactions between net atomic charges of the two bases, Era is the energy of the interaction between net charges of the atoms of one base and the dipoles induced thereby in the other base, the other symbols being the same as before.The most difficult task is to calculate the total net charges - that is to determine the
s- and p-electron densities at individual atoms of the bases. Used for this purpose are approximate quantum-mechanical calculations which are essentially modifications of molecular orbital theories. Figure 7-10 illustrates the calculated distribution of partial charges for nucleic acid bases; the presented values agree well with the known experimental data.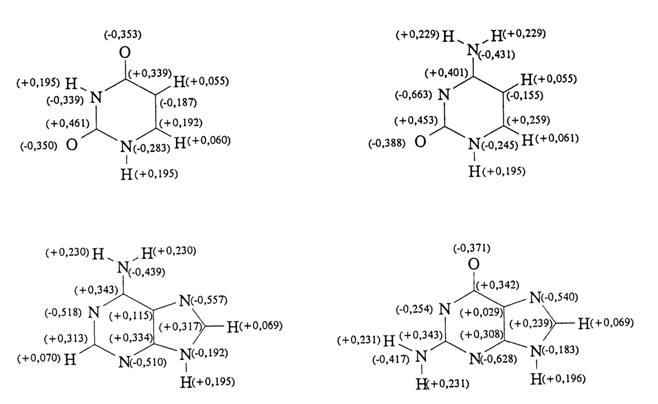
Fig. 7-10. Distribution of calculated total net charges in the nucleic acid bases. The net charges are expressed in fractions of one electronic charge (adapted from B. Pullmann and A. Saran, Prog. Nucleic. Acid Res. Mol .Biol., 18, 215-322, 1976).
7.2.1 Complementary Interactions
Such interactions were postulated for the first time by Watson and Crick. They had arrived at the conclusion that adenine in one DNA strand interacts specifically with the coplanar thymine in the other strand; guanine forms a specific pair with cytosine (Fig. 7-11).
This rule of formation of complementary pairs of bases is one of the
fundamental principles of organization of the living matter. It is realized not only
during formation of macromolecules of nucleic acids but also during their biosynthesis and
protein synthesis in the cell. A better insight into the structure of complementary
complexes of bases has become a possibility by virtue of the remarkable capacity of
alkylated derivatives of purine and pyrimidine bases ![]() to form specific pairs in the solid state.
Single crystals of such complexes have been subjected to X-ray structural analysis.
to form specific pairs in the solid state.
Single crystals of such complexes have been subjected to X-ray structural analysis.

Fig. 7-11. Structure of Watson-Crick complementary base pairs.
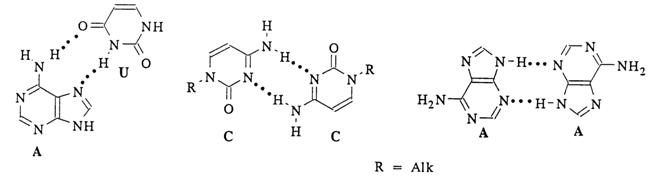
Fig. 7-12. Non-Watson-Crick mechanism of base pairing.
It has been established that adenine derivatives form pairs with those of thymine (or uracil), involving N7 of the imidazole ring (so-called Hoogsteen pairs) (Fig. 7-12). The adenine-thymine (uracil) pairs of the Watson-Crick type occurring in the DNA molecule have never been observed in crystals of monomer units.
An important property of bases (and their derivatives) is their ability to form homoassociates, such as A.A and C.C pairs.
Formation of specific complementary base pairs is also observable in organic solvents (which is impossible in aqueous solvents because water forms strong hydrogen bonds with bases) by IR and NMR spectroscopy.
What is actually observed in the former case is the shift of the absorption bands corresponding to symmetrical and asymmetrical stretching vibrations of the NH and NH2 groups of the bases at 3500-2800 cm-1. Consider 1-cyclohexyluracil (I) and 9-ethyladenine (II) as examples.
In dilute chloroform solutions, the NH group in compound (I) is characterized by a band at 3395 cm-1, while the NH2 group in compound (11) is characterized by bands at 3527 and 3416 cm-1. At such concentrations of compounds (1) and (II), no aggregates are formed. But if solutions of (I) and (II) are mixed, bands at 3490 and 3330 cm 1 appear in the IR spectrum of the mixture. The band shift is indicative of a complementary pair (I) (II) in solution as well as of involvement of the NH group of compound (I) and NH2 group of compound (II) in hydrogen bonding. The composition of the resulting complex can be studied further by looking at the relationship between the intensity of the bands corresponding to the complex and the ratio of (I) to (II) in the solution (Fig. 7-13). It can be seen that the peak intensity of the band corresponds to a stoichiometric ratio of 1:1 between (I) and (II).
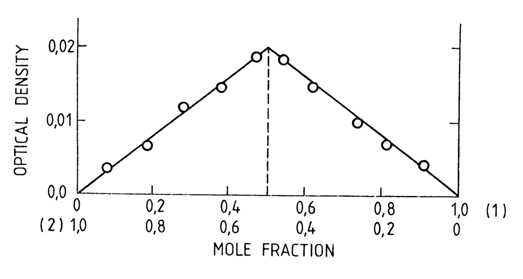
Fig. 7-13. Optical density of a 1-cyclohexyl-uracil (1) and 9-ethyladenine (2) mixture at 3260 cm-1 versus its composition (in CDC13).
Table 7-1. Total hydrogen-bonding ineraction energy in some base pairs (given in kcal/mole).

The reason why NMR spectroscopy is used to study the formation of complementary base pairs is that the signals produced by the protons of the NH groups in guanine and uracil and those of the NH2 groups in adenine and cytosine, if these groups participate in hydrogen bonding, shift toward low fields (Fig. 7-14).
The bulk of X-ray structural, IR and NMR spectroscopy data suggests that with heterocyclic bases occurring in any combination, the complementary pairing of A with T (U) and G with C is more advantageous because corresponding to this phenomenon is minimal energy of Van der Waals-London interaction, as compared to others. The results of calculating this total energy and its contributions (see above), based on net charges of the base atoms (see Fig. 7-10) are listed below in Table 7-1.
These results also indicate that the main stabilizing factors during complementary base pairing are forces of the electrostatic interactions between net charges of the atoms of both bases.
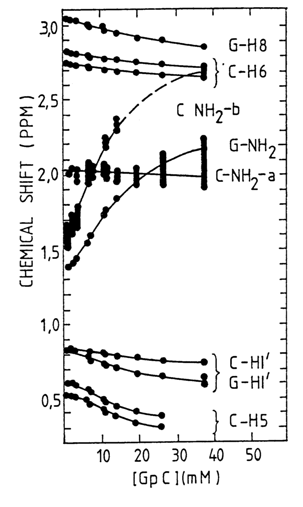
Fig. 7-14. The concentration dependence of the chemical shifts of the self-complementary ribonucleoside monophosphate GpC in aqueous solution (400 C). Compare the behavior of an amino proton of G and one of the amino protons of C with that of protons at C6 and C8 of C and G, respectively (data from T. R. Krugh et al., Biochemistry, 15, 1224, 1976).
7.2.2 Stacking Interactions
In aqueous solutions, heterocyclic base, nucleoside and nucleotide derivatives form stacked complexes in which monomer units are organized into stacktype structures whose base planes are arranged one above another. The formation of associates is due to the fact that in the base (nucleoside)-water system there arise forces precluding contacts between the nonpolar (hydrophobic) base and water. The bases come closer together, their planes overlap, and the resulting conformation is stabilized by Van der Waals-London forces acting between the adjacent bases in a stack.
Thus, we are dealing here with a typical example of a structure resulting from hydrophobic interactions.
The advantage of formation of stack-like associates of nucleic acid bases in water from the energetic standpoint is determined by a number of stabilizing factors. One of the most important ones is believed to be a change in free energy,
DF, due to reduction in size of the base surface interacting with water molecules; DF is first of all dependent on the amount of decrease in the surface tension of water as well as on changes in the degree of order displayed by the water molecules surrounding the base (i.e., on the entropy factor).Associations of bases and nucleosides can be observed in their concentrated (almost saturated) aqueous solutions and assessed by measuring the osmotic coefficient
F. It should be remembered that if the coefficient F of a substance decreases inversely with its concentration in the solution, this is a direct indication of aggregate formation. By measuring the relationship between F and concentration one can calculate the apparent equilibrium association constants K: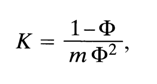
where m is the molal concentration of the substance.
The values of
F, K as well as polarizability a and dipole moments m for some nucleosides are listed in Table 7-2.Table 7-2. Parameters of stacking of nucleosides in water.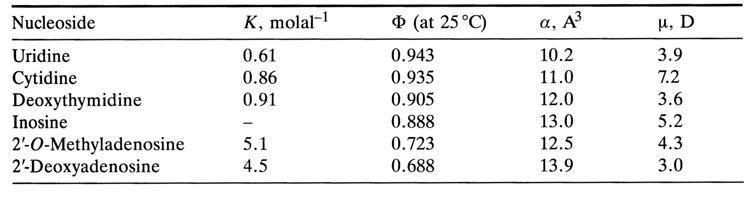
Two extremely important conclusions follow from these data:
F are lower at the same molal concentration).(1) The stacking of purine bases (and their derivatives) is more pronounced than that of pyrimidine ones (the values of K are higher and those of (
(2) Electrostatic dipole-dipole interactions are not essential to stabilize the bases in a stack (bases with higher values of
m are associated to a smaller degree); the stacking becomes more pronounced with greater polarizability a of the bases; that is, dipole (monopole)-induced dipole interactions and dispersion forces stabilize stack-like conformations substantially.Attempts have been made to more accurately describe the mutual arrangement of bases in associates. For example, by studying the relationship between chemical shifts and concentration for various protons of heterocyclic bases and their derivatives, one can find out how closely the base atoms are brought together within a stack. The following model describing the interaction between two adenines within a dimer of adenosine 5'-phosphate (in D2O) has been derived on the basis of the temperature dependence of the chemical shifts of the base protons in the NMR spectrum (this dependence has turned out to be significant only for the proton at C2) (Fig. 7-15A).
The strong tendency of purine nucleotides toward formation of structures with marked overlapping of heterocyclic bases can be illustrated by yet another example.
In experiments with the hexanucleotide GmpApApYpAp
Yp, which is a fragment of tRNAPhe from yeast and contains the minor nucleoside Y, high-resolution NMR spectroscopy revealed that the plane of the base in the nucleoside Y overlaps significantly with those of adenines in the adjacent nucleotides. And if the base in Y is removed by mild acid hydrolysis, these the adenines fill the gap formed. As a result, the stacking conformation is maintained over the entire length of the oligonueleotide and it has a structure stable even at 200C.The formation of stack-like structures is also observed in studying crystals of base, nucleoside, nucleotide and polynucleotide derivatives, however, the observations in this case include less complete overlapping of base planes, as compared to solution. Moreover, the crystal structures are marked by the ring plane of one base coming close to the polar groups of the other base, as is the case, for example, with crystals of deoxyadenosine monohydrate (Fig. 715B).
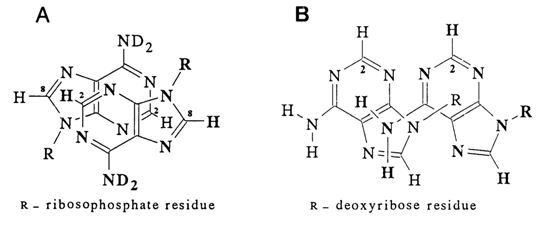
Fig. 7-15. Structure of adenosine associations in solution (A) and crystalline state (B).
Thus, when crystalline associates are formed, the forces of a dipole (monopole)-induced dipole-like interaction gain significance.