![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
6.5 DNA Sequencing by Polymerase Copying Method (Enzymatic Sequencing Method)
The polymerase method (often called enzymatic sequencing method, as opposed to chemical) was virtually the first tool proposed for sequencing large DNAS. It is based on the work done by F. Sanger, who spent more than ten years after the determination of the primary structure of insulin (1963) elaborating nucleic acid sequencing techniques. Having developed successful sequencing procedures for small oligonucleotides, Sanger's laboratory came up with what became known as the "plus and minus method" (1975). It took a mere two years for Sanger to published, along with the presentation of the chemical method (1977), a description of his modified method (DNA sequencing using chain terminating inhibitors), which became known as the "dideoxy method". This sequencing technique, based on template-directed and primed synthesis of single-stranded DNA with the aid of DNA polymerase, provides a reliable and simple way to sequence DNA, along with the above-described Maxam-Gilbert method. The potential of the method was obvious already in 1976, when Sanger and coworkers determined the primary structure of the genome of phage
FX174. Although the genome contained "only" 5375 nucleotides, the possibility to decode it completely and thus gain access to all the genetic information contained in the genome marked a breakthrough in this field. It is precisely this phage that demonstrated the possibility of encoding a gene within a gene.6.5.1 Basic Principle of the Method: Priming and Termination of Enzymatic Synthesis of DNA Copies
Conceptually, both sequencing methods - chemical and enzymatic - are identical. However, if the Maxam-Gilbert method is based on specific DNA cleavage determined by the nature of the constituent bases, the underlying principle of Sanger's method is statistical DNA synthesis terminating at one of the four nucleotides. Thus, both methods are aimed at obtaining a complete (statistical) set of DNA fragments terminating at each of the four nucleotides. It was found that synthesis with subsequent PAGE and autoradiography provides an effective means for producing such sets, as opposed to degradation.
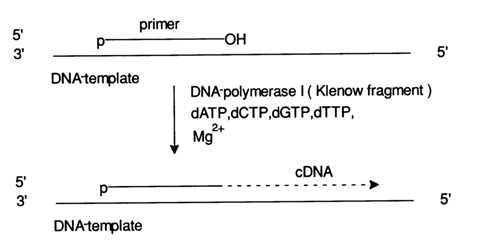
Fig. 6-11. Synthesis of a DNA (cDNA) copy using a single-stranded template DNA with a primer, which is an oligonucleotide complementary to a particular site, in the presence of all four deoxynucleoside 5'-triphosphates. The synthesis is catalysed by DNA polymerase 1, or Klenow fragment, lacking 5®3' exonuclease activity.
The substrate in the polymerase copying method is a single-stranded fragment, or a DNA template (which is essentially the molecule of interest), annealed to a short complementary fragment, or primer. Figure 6-11 shows schematically the standard procedure for copy DNA (cDNA) synthesis in the presence of Klenow fragment (DNA polymerase I), illustrating the enzymatic process underlying the method. The chemical reaction of enzymatic synthesis of internucleotide linkages, as part of cDNA synthesis, boils down to phosphorylation of the primer's 3'-hydroxyl in the template-primer complex with the next complementary deoxynucleoside 5'-triphosphate. The a-phosphate group of the latter in such an enzyme-substrate complex is attacked by the 3'-hydroxyl of the primer after departure of the pyrophosphate group (see Fig. 6-12).
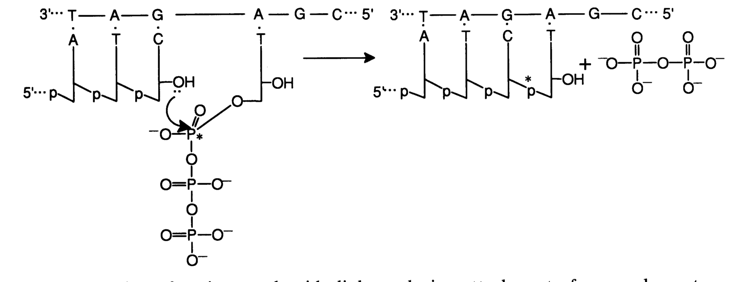
Fig. 6-12. Formation of an internucleotide linkage during attachment of a complementary nucleotide (dpT) in polymerase copying. If nucleoside 5-triphosphate 32P-labeled at the
a-phosphate group is used in the reaction, the cDNA receives a label instrumental in identifying the synthesized polynucleotide by autoradiography. dNTP 35S-labeled at the a-phosphate can also be used. In this case, bands on the autoradiogram are more distinct (because of the b-particle emission energy). However, the specific activity is usually lower and the gel has to be exposed over a longer period of time.The motive force of the copying process is formation of a productive enzyme-substrate complex: template-primer-complementary deoxynucleoside 5'-triphosphate-DNA polymerase. Naturally, with the lack of structural consistency in such an enzyme-substrate complex, no internucleotide linkage is formed; consequently, the polymerization (cDNA formation) is inhibited. It is precisely this factor that is used in sequencing by the polymerase copying method, when employed as a polymerization inhibitor is one of the four nucleoside 5'-triphosphates - the one at which copying must be partially (statistically) stopped. For this purpose analogues of ordinary dNTP substrates without a hydroxyl group at the 3' position are used i.e., 2',3'-dideoxyribonucleoside 5'-triphosphates, abridged as ddNTP):
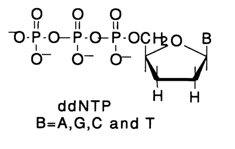
The primer is extended in the presence of all the four usual deoxyribonucleoside 5'-triphosphates and the 2',3'-dideoxy analogue (ddNTP) of one of them.
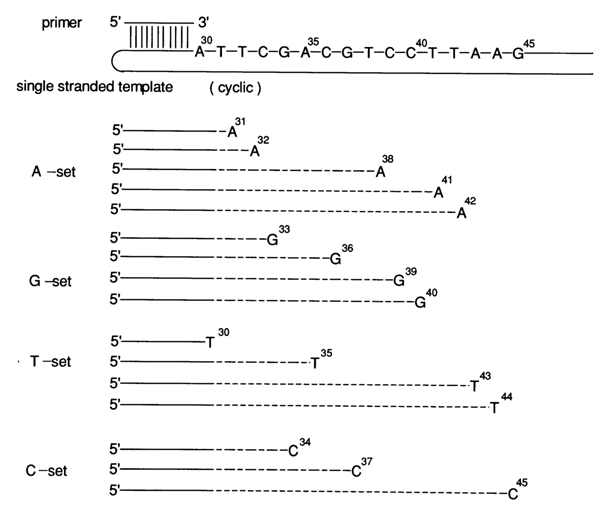
Fig. 6-13. The basic principle of chain termination during DNA sequencing by the polymerase copying method. When such a substrate as, for example, ddATP is introduced into the reaction mixture along with the usual DNTP ones, the synthesis of some chains discontinues at positions T in the template. This gives an A set consisting of all possible oligonucleotides terminating in A (positions 31, 32 and 38 corresponding to T in the template). When ddGTP, ddTTP and ddCTP substrates are used, G, T and C sets are obtained, respectively, the numerals indicating the corresponding chain-terminating nucleotides.
The analogue is inserted into the growing strand, the enzyme recognizes it, and another internucleotide linkage is formed with its participation. However, further extension of the complementary strand complex in this position is impossible (see Fig. 6-12) because the inserted analogue lacks a 3'-hydroxyl group (polymerase substrate). One of the four analogues is used to obtain a corresponding set of template copying products having a common origin, which is the primer, and terminating at a nucleotide of appropriate type.
Figure 6-13 is a schematic demonstration of this sequencing procedure, from which it becomes evident that the polymerization in four simultaneous reactions, each involving statistical insertion of the corresponding dideoxynucleoside 5'-phosphate, yields four sets of oligonucleotides (A, G, C an T sets). Each such set comprises oligonucleotides terminating at the 3' end with a nucleotide (A, G, T or C) which is complementary to its counterpart in the template. If a single dNTP with a 32P-labeled a-phosphate group (usually a32P-ATP) is introduced into a reaction mixture, all oligonucleotides in the A, G, T and C sets become labeled. The separation of these sets in PAGE by length makes it possible, just as in chemical sequencing, to read the initial sequence from the band pattern on the autoradiogram. Figure 6-14 is an autoradiogram of the reaction mixtures (A, G, T and C sets). In order to obtain a good "statistical" set of cDNA, one must vary the ratio between the terminating and the corresponding ordinary nucleoside 5'-triphosphates. At a dNTP/ddNTP ratio equal to 1:100, it is possible to obtain a well resolved set of bands and read as many as 200 and more nucleotides. A lower ddNTP concentration gives rise to larger copying products, which, in combination with a lower concentration of acrylamide, increases the number of nucleotides that can be read off the same gel slab to 300.
In addition to 2',3-dideoxynucleoside 5'-triphosphates, the corresponding arabinonucleoside 5'-triphosphates can be used as terminators of enzymatic synthesis of cDNA, such as
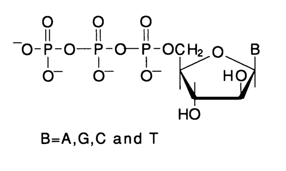
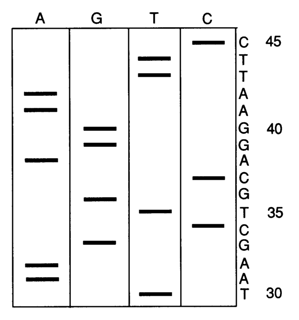
Fig. 6-14. Autoradiogram showing the A, G, T and C sets of Fig. 6-13. The cDNA sequence read off on the right begins after the primer or, in other words, at position 30 and ends at position 45 from the 5' end. This allows one to write the complementary sequence or the primary structure of the template between nucleotides 30 and 45 from the 3' end:
30
35
40
45
3'... ATTCGACGTCCRTTAAG... 5'
which are inserted into the growing cDNA strand but inhibit subsequent formation of internucleotide linkage. The inability of "arabinose" fragments to participate in the latter seems to stem from the unfavorable conformation of the 3'-hydroxyl group. Perhaps, this also has to do with the configuration of the 2'-hydroxyl. An alternative type of terminators includes 3'-azido derivatives of 2'-deoxyribonucleoside 5'-triphosphates, introduced into sequencing by A. A. Kraevsky and coworkers:
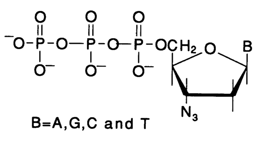
Naturally, termination in this case involves an azido group, instead of the 3'hydroxyl, in the substrate of DNA polymerase 1. Interestingly, insertion of such modified nucleotides into the strand takes place none the less. Important sites of recognition by this enzyme of its substrates include the 5'-triphosphate group and the heterocyclic base. The primers in the polymerase copying method are either single-stranded synthetic oligonucleotides or the restriction fragments of the DNA under analysis (i.e., fragments resulting from hydrolysis of the DNA under analysis with a specific restriction endonuclease).
The polymerase copying method is commonly used to determine reliably anywhere from 15 to 200 (and sometimes more) nucleotides beginning from the primer. Figure 6-15 is an autoradiogram of a typical sequencing gel, produced by the enzymatic method. The primary DNA structure is represented by the shown sequence. For good separation of fragments ranging in size
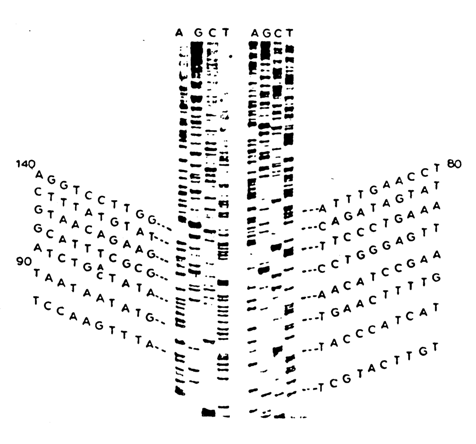
Fig 6-15. Autoradiogram of a sequencing gel produced by the enzymatic method. Written on either side of the sequence of nucleotides read off from this autoradiogram.
from 50 to 200 nucleotides a 8 % polyacrylamide gel is used. In the case of smaller fragments with less than 100 nucleotides, 12 % polyacrylamide gel is the right choice. On the other hand, reliable separation of larger fragments (with more than 250 nucleotides) is achieved with the aid of a 6 % gel.
6.5.2 Strategy for Sequencing Large Single-Stranded DNAs by the Enzymatic Method
The enzymatic dideoxy sequencing method makes it possible to determine the primary structure of any DNA that can be obtained in a single-stranded form. There are several procedures for preparing single-stranded DNAs, cloning in phage M13 being the most widely used.
Any double-stranded DNA fragment, irrespective of its primary structure, can be inserted into a vector molecule to obtain a recombinant DNA. Separation of DNA fragment mixtures is performed using the cloning technique. A mixture of fragments of any degree of complexity can be separated by plating bacteria transformed by a mixture of recombinant DNAs. In order to find all fragments of the starting mixture, every clone must be used.
To simplify the clone screening procedure, vectors have been developed with a special structure permitting one to distinguish between bacterial colonies carrying a vector with an insert and an "empty" vector by the color of the colonies in plates with an indicator medium. The first such vectors were developed for producing single-stranded DNAs or, in other words, templates for the enzymatic sequencing method. Figure 6-16 illustrates the basic principle of using vector M13 (replicative form of phage M13 DNA) to separate the strands of any DNA fragment.
The starting fragment is incorporated into the replicative double-stranded form of M13 DNA at the sticky ends of the EcoRI site occupying a functionally insignificant portion of the phage. Two orientations are possible for inserting a fragment. The prepared mixture of molecules is used to transform a bacterial cell and isolate a single-stranded form from the mature phage DNA, containing either strand A or strand B of the starting DNA fragment. Used for the sequencing of such DNA fragments (A or B) is the same short synthesized or isolated (with the aid of restriction endonucleases) primer forming a complex with the vector DNA near the site of insertion of the starting DNA (see Fig. 6-17).
Here is a brief description of the vector - phage M13 - used for obtaining, in a simple procedure, single-stranded DNAs ready for sequencing. M13 is a filamentous coliphage having a single-stranded genome with a relative molecular weight of 2.106. After inoculation with E. coli the single-stranded DNA becomes double-stranded, or converted into a replicative form, and during amplification produces about 300 copies per cell. The inoculated cells are not lysed but continue to grow slowly, discharging the phage into the medium.
The filamentous phage M13, just as some other analogous phages (fl, fd), are characterized by three factors extremely important for use in DNA cloning, namely:
(a) the phage DNA can be isolated as a double-stranded molecule;
(b) the DNA "harbored" by the phage may be longer than the phage DNA;
(c) the phage does not lyse the host cell (as opposed, for example, to the lambda phage).
The M13 genome has a small region (about 100 b.p.) which, as has already been mentioned, is functionally insignificant for development of the phage; it is precisely into this region that the foreign DNA can be inserted. Phage M13mp2, for example, has the following structure making it a suitable vector. The above region receives a lac promoter and a fragment of the lacZ gene encoding the first 146 amino acids of the enzyme
b-galactosidase, so that the N-terminal fragment of this protein is synthesized in the E. coli cells inoculated with phage M13mp2. If use is made in this case of the E. coli strain gal having mutations at the lacZ gene segment in question and, therefore, syn-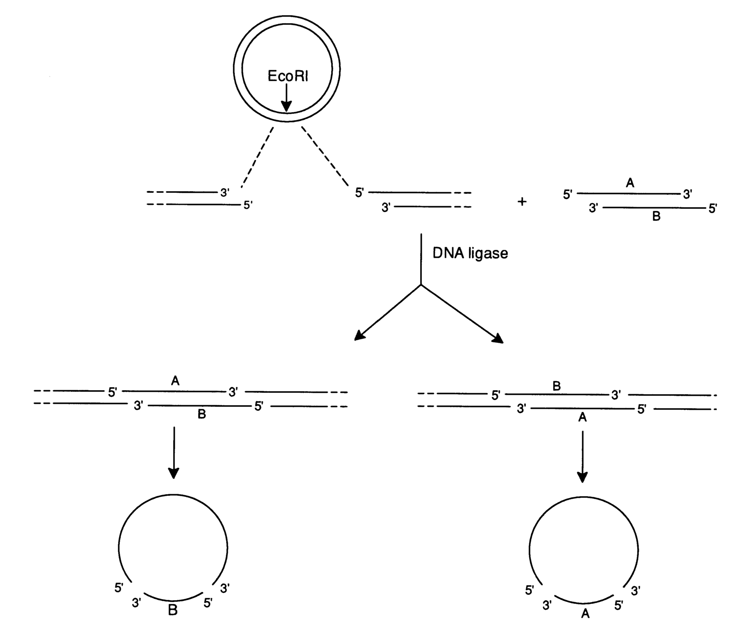
Fig. 6-16. Separation of DNA strands (A and B) by cloning in a vector based on the filamentous bacteriophage M13.
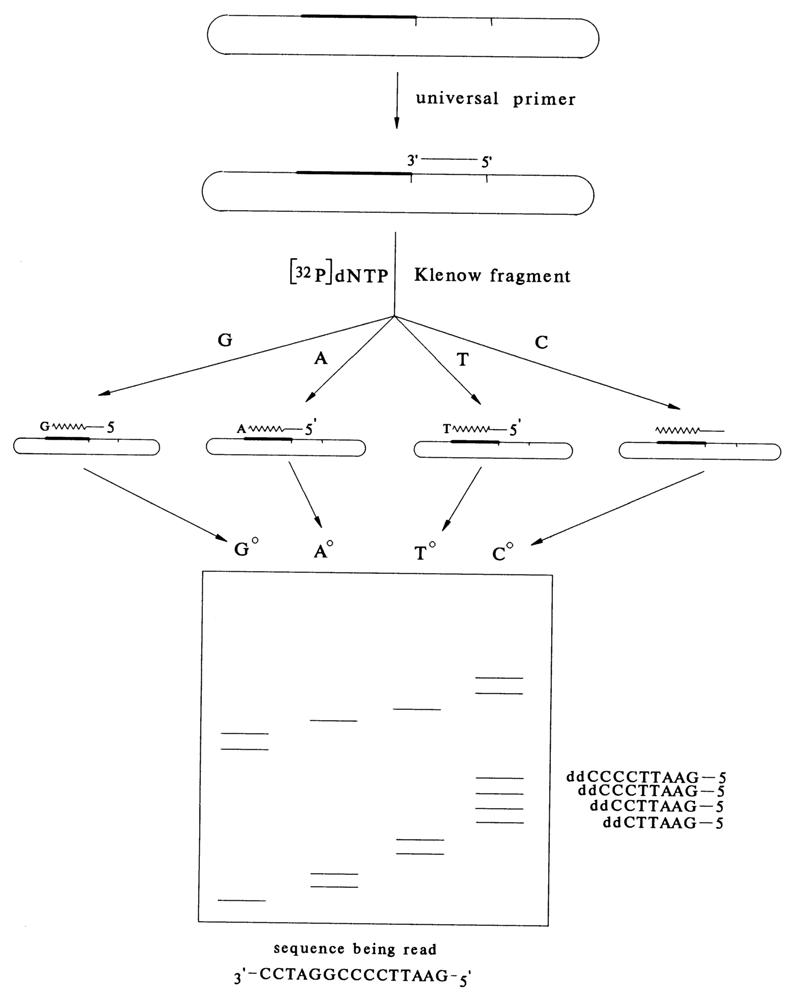
Fig.6-17. Sequencing of a single-stranded DNA fragment in phage M13 DNA with the aid of a universal primer.
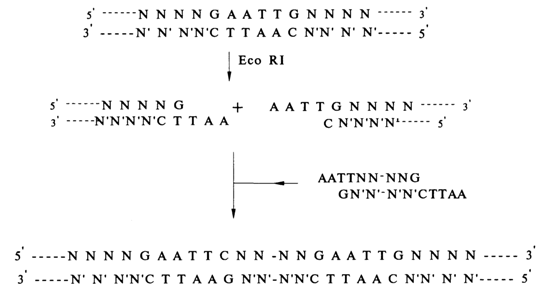
Fig. 6-18. Insertion of a foreign DNA at the EcoRI site of M13.
the sizing inactive b-galactosidase, its inoculation with the phage produces cells of the gal+ phenotype, capable of hydrolysing b-galactosides. This is a result of association of the mutant enzyme encoded by the bacterial DNA with its complete N-terminal fragment encoded by the phage DNA, giving rise to a complex exhibiting b-galactosidase activity. The gal' cells can be easily identified because their presence leads to hydrolysis of the chromogenic b-galactosides added to the medium so that the latter turns dark blue. In M13mp2, the fifth and sixth amino acid codons of the b-galactosidase gene correspond to the EcoRI restriction endonuclease site Oust as in the subsequent generations of this vector; see, e.g., M13mp7 and other vectors in Figs. 6-18 and 6-20). Insertion of a foreign DNA (whose primary structure is to be determined) at this site of M13mp2 usually precludes the above-discussed transformation of gal- cells into gal+, and the colonies remain colorless. This serves as an indicator for selecting clones containing the phage with recombinant DNA.
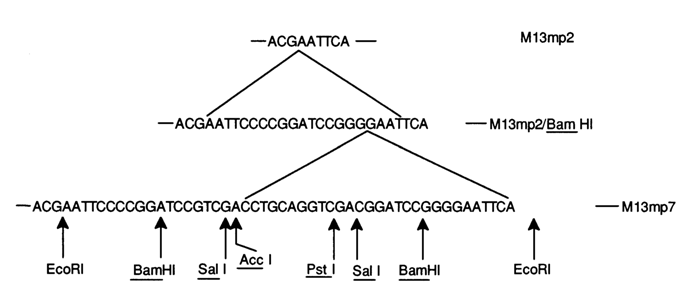
Fig. 6-19. Primary structure of a segment of vector M13mp2 near the EcoRI site and the corresponding region in vectors M13mp2/BamHI and M13mp7 constructed on its basis.
As has already been mentioned, a universal cloning vector, M13mp7, was constructed at a later date (see Fig. 6-19). To this end, inserted into the M13mp2 segment was a synthetic DNA containing unique sites of some restriction endonucleases (see Table 6-5). Listed on the left are sequences recognized by restriction endonucleases whose sites are located in vector M13mp7. The right column shows a set of enzymes capable of forming, during DNA hydrolysis, sticky ends corresponding to the sites in the left column.
Shown schematically below is a complete procedure for obtaining a singlestranded DNA suitable for enzymatic sequencing. Figure 6-20 represents the structure of the last-generation vector phage M13mpl8 with a decoded polylinker at which a foreign DNA is inserted.
Table 6-5.
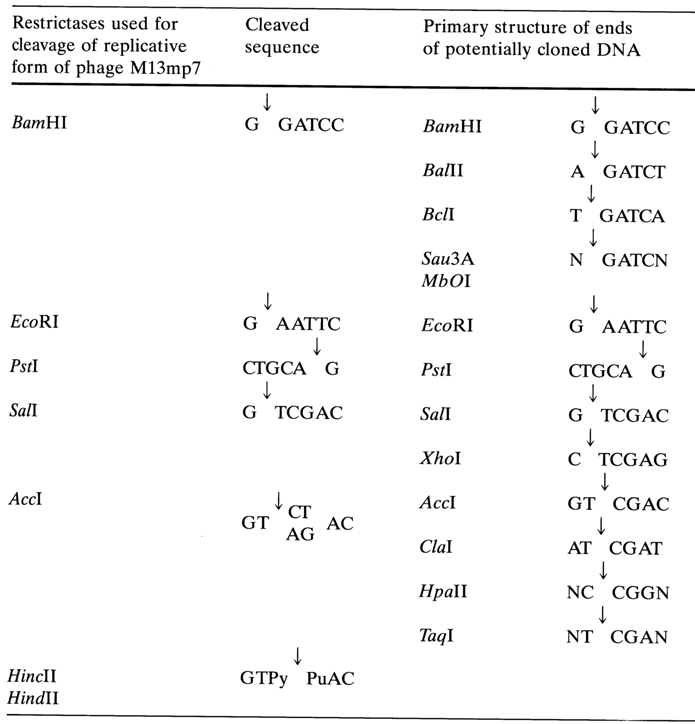
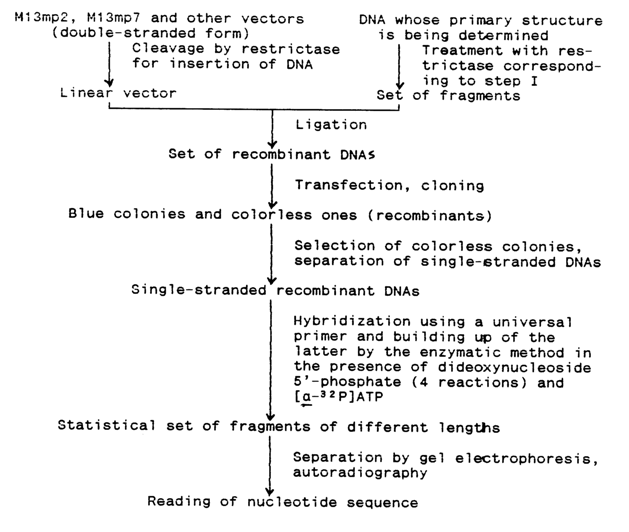
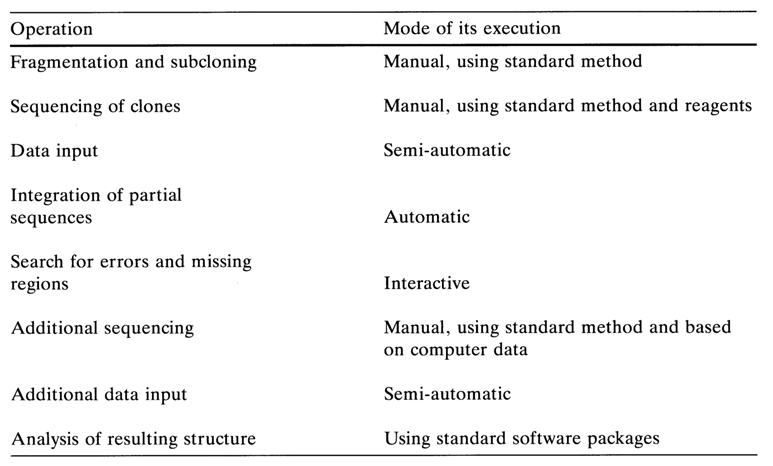
Figure 6-21 illustrates the primary structure of polylinker regions inserted into new generations of vectors based on phage M13. As a consequence of engineering of universal vectors and improvement of methods for cloning and isolation of single-stranded DNAs suitable for sequencing, determination of the primary structure of high-molecular weight DNAs is now possible without restriction endonucleases and their fragmentation can be done using such non-specific agents as DNase I or by sonication. An advantage offered by such an approach is that a restriction map of the DNA removal under analysis is no longer necessary, which simplifies the experimental procedure significantly. The sequencing involves a fraction containing fragments of limited length (up to 1000 b.p.), their mixture being inserted into the vector by way of blunt- or sticky-end ligation (in the latter case, Tinkers must be attached to the fragment ends). The result is a mixture of different recombinant DNAs structurally corresponding to the replicative form of phage M13 DNA, each containing a particular fragment of the starting DNA. Then, these DNAs are cloned into E. coli, the single-stranded phage DNAs are isolated from individual clones, and the inserts they contain are sequenced by the above-described method using a universal primer (Fig. 6-22). The results are run through a computer to reconstruct the primary structure of the DNA being investigated as a whole. Such a technique (so-called blind sequencing) is by far the fastest, ensuring a sequencing rate of 5000 nucleotides per day on the average. Table 6-6 summarizes the algorithm of this sequencing approach.
A prerequisite for such a sequencing procedure is availability of the appropriate software packages and a nucleotide sequence data base used at the data processing stage.
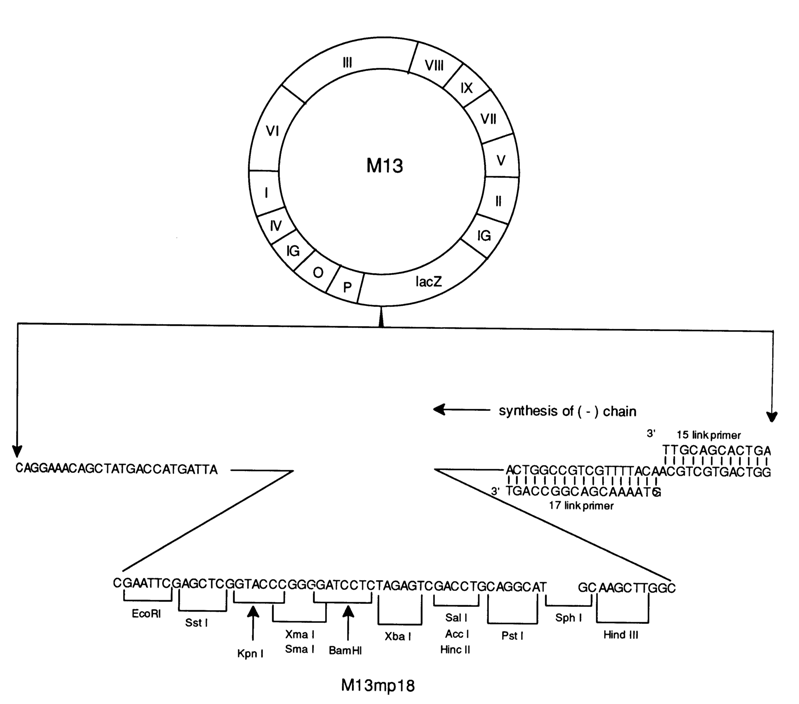
Fig. 6-20. Structure of vector M13mpl8. Shown below is the primary structure of the polylinker and the adjacent segments as well as the 15- and 17-unit primers initiating the synthesis of cDNA during sequencing by Sanger's method.

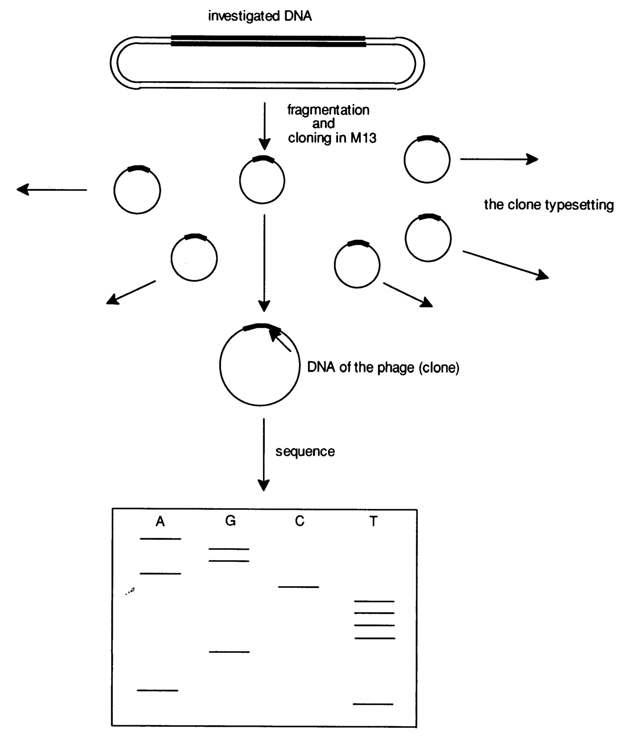
Fig. 6-22. Sequencing of high-molecular weight DNAs by the enzymatic method. Cloning in M13 of its fragments (resulting from nonspecific cleavage), separation of clones, and subsequent use of a universal primer for each clone (the primer is shown as the arrow indicating the direction in which the template is copied).
Table 6-6.