![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
6.4 Controlled Chemical DNA Cleavage Method (Chemical Sequencing)
6.4.1 Basic Principle of the Method
The method was proposed in 1977 by the American scientists A. Maxam and W Gilbert. It was immediately recognized as very simple, fast and reliable and is now most extensively used world-wide. Its simplicity is determined not only by the sequencing procedure itself, but also by the preparation of the starting individual DNA sample. All is needed, as has already been mentioned, are restriction endonucleases and enzymes enabling incorporation of label into the 5' or 3' end of DNA.
The basic principle of the method is location of each of the four bases along the polynucleotide chain relative to one (5'- or 3'-) terminal nucleotide which thus becomes a reference point. To this end, one resorts to statistical chemical modification in four different reactions involving one or two of the four DNA bases to permit subsequent quantitative cleavage of the sugarphosphate backbone at the modification sites. The ideal result is obtained when the base in question in each 200- to 300-nucleotide DNA fragment under investigation is modified only in one particular position. Cleavage of the polymer gives rise to a plurality of molecules differing in length according to the position of the base with respect to the reference point. If, for example, the reference point is a 32P-labeled 5'-terminal nucleotide, then only molecules containing the 5'-terminal sequence can be "seen". To determine the length of the resulting fragments use is made of polyacrylamide gel electrophoresis (PAGE) under denaturing conditions (in the presence of urea), when the products of partial DNA cleavage are sorted by size. When electrophoresis is over, its result is fixed by placing the gel on an X-ray film. After exposure and development, the film (autoradiograph) shows dark bands corresponding to the position of 32P-labeled oligonucleotides in the gel. Naturally, unlabeled oligonucleotides, also formed during chemical degradation, do not show on the autoradiograph. While reading the sequence out from the latter, the experimenter simply notes down the base-specific reagent that has cleaved the chain at the next nucleotide.
Shown schematically below is a sequence of nucleotides in a 20-unit polydeoxyribonucleotide and the process of degradation of the polynucleotide chain at the cytosine (C) sites, as well as the resulting labeled and unlabeled oligonucleotides.

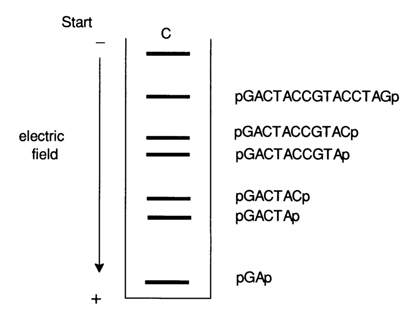
Fig.6-2. Autoradiogram of a mixture of oligonucleotides after statistical degradation of the 20-unit DNA fragment at C (see text) and separation by PAGE. Shown on the right is the primary structure of the corresponding labeled oligonucleotide.
Figure 6-2 is an autoradiogram of this oligonueleotide mixture after PAGE, used to "read" the sequence of the cytosine nucleotides along the icosanucleotide chain from the 5' end.
After such an experiment, the formula of the icosanucleotide under investigation can be written as follows:
5
10
15
20
N-N-C-N-N-C-C-N-N-N-C-C-N-N-N-C-N-N-N-N
where N is the unknown nucleoside.
The other letter symbols - A, G and T standing for the corresponding nucleotides - are arranged in the same fashion along the chain of the DNA fragment of interest.
Thus, by combining the PAGE technique with determination of radioactive phosphate A. Maxam and W Gilbert successfully developed a rapid method for determining the primary structure of DNA through its chemical degradation. It is now hard to imagine that as recently as the mid seventies it took years to determine the primary structure of a 30- to 40-nucleotide DNA fragment, whereas already by the early eighties analysis of structures containing a thousand and more nucleotides had become a matter of just a few days work.
6.4.2 Chemical Methods for Specific Cleavage of Polydeoxyribonueleotide Chain
Statistical (partial) specific chemical cleavage of internucleotide linkages in DNA at each of the four nucleotides in four different reactions conducted simultaneously forms the basis of sequencing.
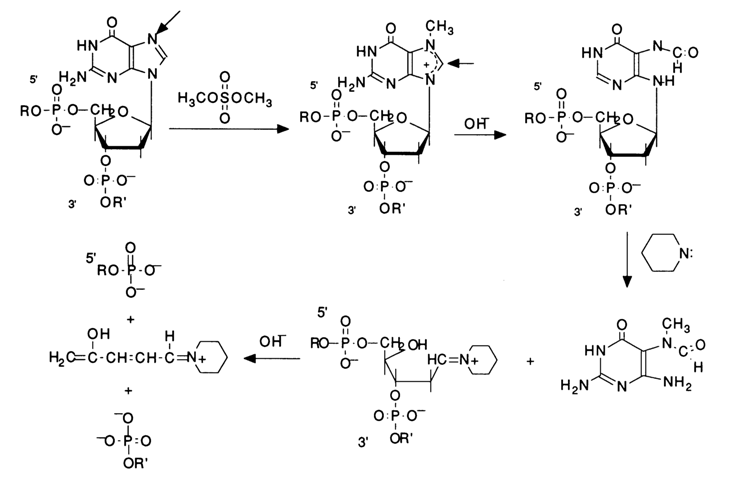
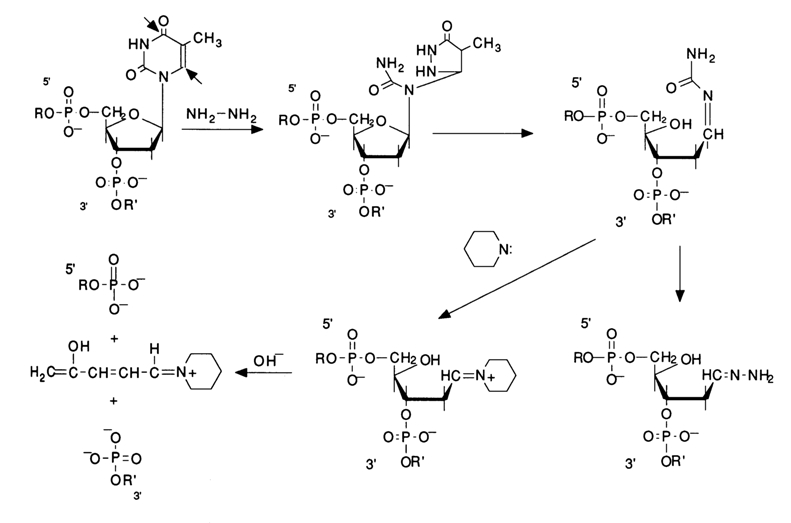
Practically, every chemical cleavage of a particular internucleotide linkage comprises three consecutive steps: modification of the heterocyclic base, detachment of the modified base from the sugar, and
b-elimination of the internucleotide phosphate groups from the 3' then 5' positions of deoxyribose of the polynucleotide which "lost" the base during the second step. Figure 6-3 illustrates these steps in the context of modification of guanines by their methylation with dimethyl sulfate, followed by cleavage of the N-glycosidic bond an b-elimination.Emphasis should be placed on the intimate realtionship between the reaction at each step of the process and the preceding ones. The cleavage of internucleotide linkages in DNA occurs only at the sugar where the base has been detached. The specificity of such degradation of the polynucleotide chain is ensured at the first step, when the base is modified, and the reaction is conducted under conditions "mild" enough to keep the degradation within the desired limits. The subsequent reactions, namely, removal of the base and
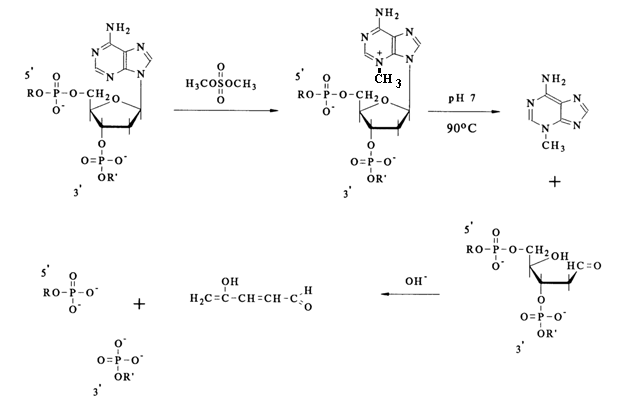
b-elimination, proceed in a quantitative manner. Hence, the crucial factor is the selection of the modifying reagents ensuring specificity of the modification (involvement of only one base in the reaction) and, consequently, the site at which the polynucleotide chain is to undergo cleavage. The main modifying agents are dimethyl sulfate and hydrazine. Dimethyl sulfate easily methylates the nitrogens of the purine bases in DNA. Methylation in doublestranded DNA proceeds perceptibly only in two positions - at N7 of guanine and N3 of adenine. It should be pointed out that the methylation of guanine in DNA proceeds at a rate almost an order of magnitude greater, as compared to adenine ones. This is precisely what makes cleavage possible during sequencing. In both cases, the methylation of purines leads to the destabilization of the N-glycosidic bond linking the base and the sugar.Given below is the scheme of guanine base methylation in an oligonucleotide chain. The positive charge emerging during methylation of N7 is delocalized in the imidazole ring among the atoms N7, C8 and N9. This weakens the N-glycosidic bond, and the latter is broken in a broad pH range at a rate several orders of magnitude greater than in the case of unsubstituted deoxyguanosine.
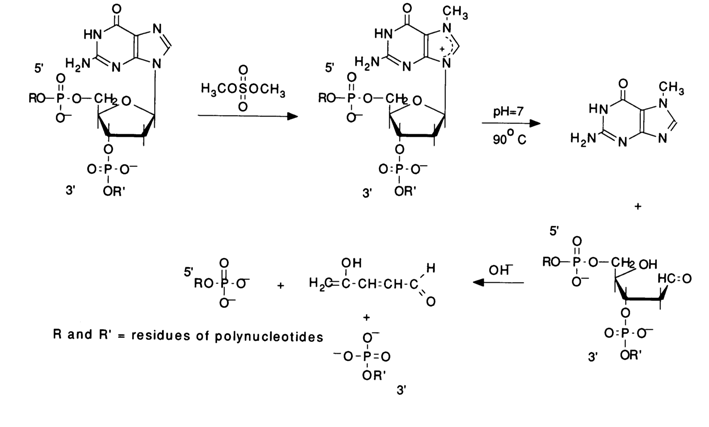
The resulting "base-free" deoxyribose readily passes from the cyclic form into an open one characterized by reactions of
b-elimination of the 3'-phosphate group. Such reactions are usually catalysed by alkalis or organic bases. The unsaturated sugar emerging after "departure" of the phosphate dianion associated with the 3'-terminal polynucleotide sequence being handled again undergoes b-elimination, but this time the 5'-phosphate group is involved. A consequence of such a double b-elimination is the degradation of the polynucleotide chain at the modified deoxyguanosine link.If such a transformation in different molecules of a polynucleotide being sequenced affects only one of the many deoxyguanosine links (the ideal case of limited modification), the reaction mixture accumulates all possible oligonucleotides - products of polymer chain cleavage at sites previously occupied by deoxyguanosine.
As has already been mentioned, the methylation of
N3 in adenines also takes place under the same conditions, albeit at a rate one order of magnitude slower, with the result that the degradation of DNA occurs at sites previously occupied by adenine nucleosides as well.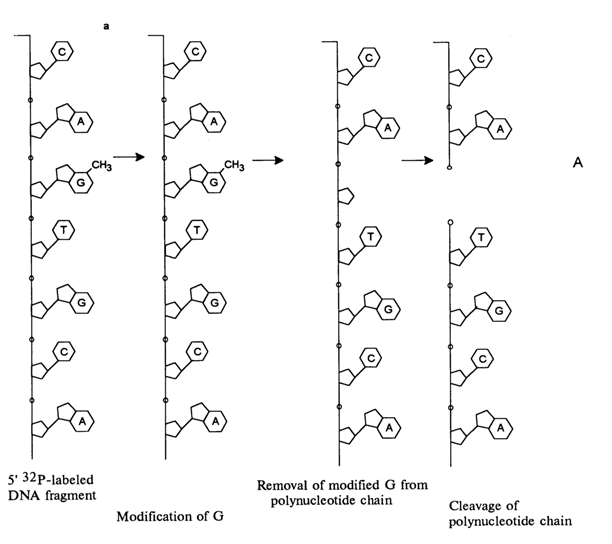
Fig. 6-3. Sequencing of 5'-labeled DNA fragment by limited (statistical) methylation with dimethyl sulfate, followed by removal of N7-methylguanine and
b-elimination of the phosphate groups linked with this nucleoside in the polynucleotide chain: (a) chemical reactions at the modification site; (b) structure of the fragments resulting from limited modification of guanines in the 18-unit oligodeoxyribonucleotide, having a 32P-labeled 5'-terminal phosphate.The positive charge emerging at N3 during the methylation of the pyrimidine fragment of the adenine ring doubles the lability of the glycosidic bond, as compared to N7-methyl deoxyguanosine. The subsequent cleavage of the polynucleotide chain at the sites of N3-methyl deoxyadenosines proceeds at neutral pH values (ca. 7), its mechanism being similar to that described above for deoxyguanosine links.
When this process is conducted on end-labeled DNA with subsequent electrophoretic separation of the degradation products and autoradiography, the positions of guanine and adenine nucleotides in the polynucleotide can be identified. However, since the methylation of N7-guanine in this reaction proceeds at a much faster rate, most DNA cleavages will occur at guanines. The autoradiograph will show that the bands corresponding to cleavage at G are always more intense than those corresponding to cleavage at A (G > A). And if the reaction of base detachment is conducted at pH 2 under conditions of much milder hydrolysis of N-glycosidic bonds in 3-methyladenine links, followed by the usual alkaline treatment, the A bands on the autoradiograph will be more intense than the G bands (A > G) (see Table 6-3). These differences in the rates of methylation and detachment of methylated purines were used to distinguish between the constituent guanines and adenines of DNA. In practical experiments, these differences did not always manifest themselves to the same degree, which is why the main efforts were aimed at providing the right conditions for locating adenines in reactions not involving guanines. One of such reactions turned out to be degradation of DNA with a 1 N alkali at 900 C, opening the adenine ring and, after subsequent treatment with piperidine, breaking the polynucleotide chain at modified A sites. These conditions are conducive to partial degradation at C sites as well (A > C). Provision for three different sets of conditions for DNA modification (G > A, A > G, A > C) has made it possible to reliably determine the positions of G and A in the polynucleotides under investigation.
Later, the right conditions for cleavage only at guanine links were found.
G-specific cleavage is conducted as follows: after methylation with dimethyl sulfate, the reaction mixture is treated with piperidine in free-base form, whose presence allows only guanines to be removed from the polynucleotide chain. It is well known that the imidazole ring in N7-methylguanine nucleoside is opened by mild alkaline treatment (pH > 10). The resulting N-glycoside formed with the participation of the primary amine is converted into azomethine which is typically involved in
b-elimination reactions, just as the aldehyde form of the depurinated deoxyribose.The G-specific reaction has made it possible to simplify the procedure of locating purine nucleotides. The G+A cleavage started being conducted in an acid medium.
Acid depurination also leads to DNA cleavage at guanine and adenine, but no distinction between the two is possible. The mechanism of acid hydrolysis of N-glycosidic bonds in purine nucleosides was treated at length elsewhere. As is currently believed, only protonation of N7 in guanine and N3 in adenine renders the N-glycosidic bond labile. The corresponding bonds in the pyrimidine links of DNA at low pH values (<5) are much more stable. In the latest version of sequencing (A+G modification), limited hydrolysis with formic acid is used.
To determine the position of pyrimidine bases in the polynucleotide chain, they are modified with hydrazine. The most reactive fragment in pyrimidines of nucleic acids is the double bond C5-C6. In a reaction with hydrazine, one of its molecules is added at the above-mentioned double bond with the result that the strong nucleophilic group of hydrazine becomes linked to the most electrophilic carbon C6.
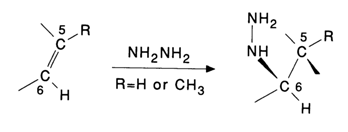
The pyrimidine ring is no longer planar and loses its aromatic properties. Notably, the presence of the methyl group at position 5 (thymine) enhances the stability of this pyrimidine base toward nucleophiles. This is why thymines enter into such reactions with greater difficulty as a rule. Naturally, such transformations are unusual for purine bases because the double C-C bond in the purine nucleus, corresponding to C5-C6 in pyrimidines, is also included in the aromatic system of imidazole.
After addition of the hydrazine molecule at the double bond C5-C6, the heterocyclic ring of pyrimidines is opened as a result of intramolecular nucleophilic substitution involving the added hydrazine, for example:
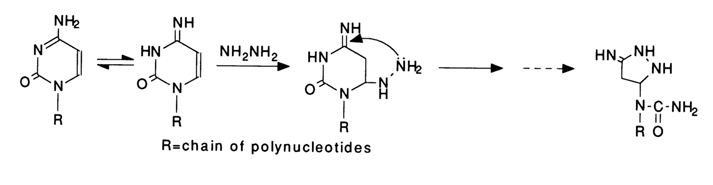
The new five-membered ring now includes the hydrazine nitrogens, whereas those of cytosine are beyond the ring. These and subsequent transformations of one of the cytosines in the polynucleotide chain are shown below.
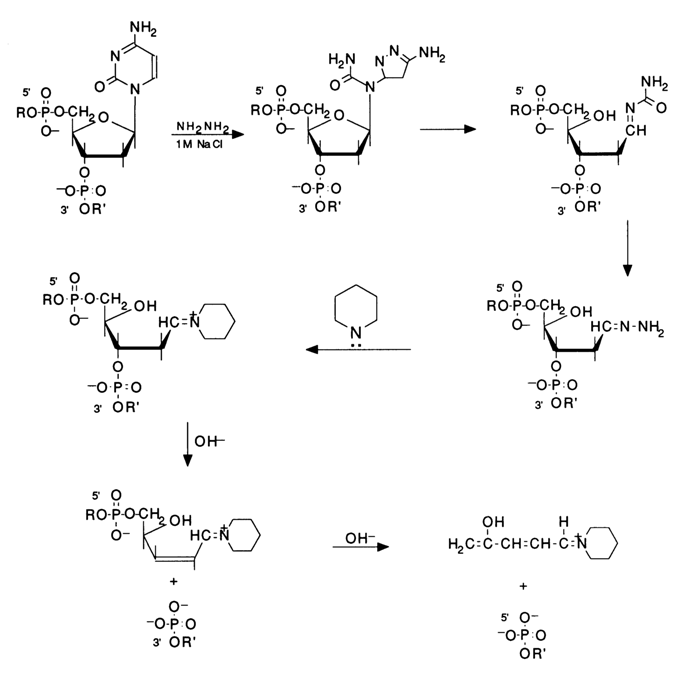
The available data on the mechanism of the reactions between pyrimidine bases and hydrazine in nucleic acids suggest that one of the following groups may remain linked to the sugar at the modification site: the glycosidic nitrogen of urea in the form of a secondary amine; the glycosidic nitrogen in the form of tertiary amine; or hydrazine itself. In all structures of this type the bond linking the sugar to the nitrogen is sufficiently reactive. Treatment with piperidine easily gives the corresponding azomethine with a fixed positive charge, characterized by
b-elimination of the 3'- then 5'-phosphate, described above in the context of structures with sugar in an open (aldehyde) form, resulting from depurination. The above-described mechanism of DNA cleavage at cytosines is still hypothetical and needs additional studies. However, the experimental evidence available so far (hydrazinolysis with 17-18 M hydrazine followed by treatment with 1 M piperidine, 900 C, 30 min.) indicates that the right conditions for cleavage of the internucleotide linkages in the cytidines of the polynucleotide chain have been chosen.The modification of thymines in DNA with hydrazine proceeds in a similar fashion:
In this case, too, the treatment of the DNA modified with piperidine at T is followed by its degradation at the same units (the same
b-elimination mechanism is involved).Thus, the above conditions are conducive to modification and cleavage of the polynucleotide chain at both pyrimidines (modification at C+T). By accident, while refining the method, A. Maxam noticed that addition of a salt (1 M NaCI) to the experimental solution drastically slows down the reactions at thymines. This finding became useful in elaborating a method for specific DNA cleavage only at cytidines (modification at C) .
All these studies into statistical chemical modification and degradation of DNA have resulted in an optimal procedure for specific cleavage of the polynucleotide chain during sequencing, namely: (G); (G+A); (C+T); (C). The advantages of this procedure are as follows.
Firstly, all of the four reactions involve piperidine (for breaking the sugarphosphate backbone), which is removed by evaporation, while the salt-free cleaved DNA can be very easily dissolved in a few microliters of formamide for application on a thin (0.3 mm) slab gel for sequencing.
Secondly, the side processes normally occurring during the degradation have been minimized in the four reactions, which ensures good separation during PAGE and permits hundreds of nucleotides to be sequenced from the labeled end of DNA by this method.
Thirdly, the sequencing scheme (G); (G+A); (C+T); (C) makes the autoradiograph rather easy to "read" by virtue of its inherent 1:2:2:1 symmetry (see Fig. 6-7). The central columns being flanked on both sides by the G+A and C+T ones each containing a base from the corresponding adjacent pair makes the sequencing picture most vivid.
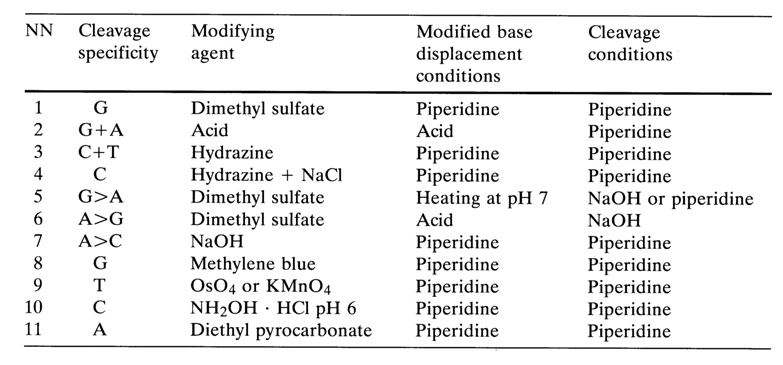
None the less, Table 6-3 below lists the main base-specific DNA cleavage reactions used in sequencing.
Reactions 5 through 11 are conducted only when it becomes necessary to verify the results obtained the usual way.
6.4.3 Obtaining Individual DNA Fragments and 32P-Labeling
The starting material for sequencing usually includes certain duplex DNA fragments isolated after treatment of linear DNAs with restriction endonucleases or similar restriction of a cyclic DNA (from plasmids, phages, viruses) at a single site. What is more, if the DNA duplex is long enough and contains, say, more than a thousand nucleotide pairs, it must be broken into shorter fragments because in a single sequencing experiment only 200 to 300 nucleotides can be handled. To this end, the restriction sites are mapped and DNA is cleaved at these sites. Such procedures result in double-stranded DNAs with unique terminal sequences (see, e.g., Tables 6-1 and 6-2) which can be labeled at the 5' or 3' end, depending on which end of the strand is important for sequencing. In each case, the end-labeled sample represents a duplex DNA. Since only one labeled strand is necessary for analysis, the next step is either separation of the strands (Fig. 6-4a) or treatment with a restriction nuclease with a specificity different from the initial one (Fig. 6-4b).
The state-of-the-art cloning techniques using plasmids and phages as vectors make it possible to obtain any DNA fragments in amounts of hundreds of picomoles, which is quite sufficient for sequencing of a chain up to five thousand base pairs long.
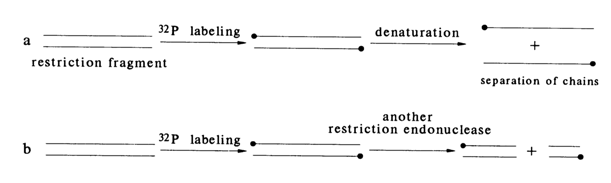
Fig. 6-4. Two ways of preparing DNA fragments with a single label: (a) DNA labeled at two strands is subjected, after denaturation, to PAGE for separation of the strands; (b) DNA labeled at two strands is cleaved by a second restrictase (E) to give duplexes labeled at one strand only.
As has already been mentioned, one of the basic principles of the chemical sequencing method is the presence, at the 5' or 3' end of the DNA under analysis, of an atom or a group of atoms that would enable detection of the chemical degradation products after the separation. More often than not, a 32P-phospate group or 32P-labeled nucleotide incorporated into the DNA with the aid of appropriate enzymes is used for this purpose. The labeled terminal nucleotide must be the same in all molecules of the DNA under investigation. Since fragments obtained by controlled cleavage of a native DNA using restriction endonucleases are most commonly involved in the sequencing of naturally occurring DNAs, the terminal nucleotides are identified by the specificity of the corresponding enzyme. To incorporate the label use is made of three enzymes and three different 32P-precursors:
Labeling the 5' Ends. For direct phosphorylation of the ends of restriction fragments to take place, their 5'-phosphate must be removed in advance with the aid of alkaline phosphatase. Double-stranded DNAs with protruding 5' ends (see Table 6-2) are more readily phosphorylated by polynucleotide kinase, as compared to DNAs with blunt ends or a protruding 3' end. Table 6-4 lists some of the restriction endonucleases which leave protruding 5' ends after DNA cleavage.
Figure 6-5 shows schematically the procedure of labeling double-stranded fragments at the 5' ends. After dephosphorylation, alkaline phosphatase is removed by way of phenolic extraction and the precipitated DNA is treated with a polynucleotide kinase and [
g-32P] ATP with a specific activity of 1000 Ci/mole.Table 6-4. Restriction Endonucleases Cleaving DNA into Fragments with a Protruding 5' End
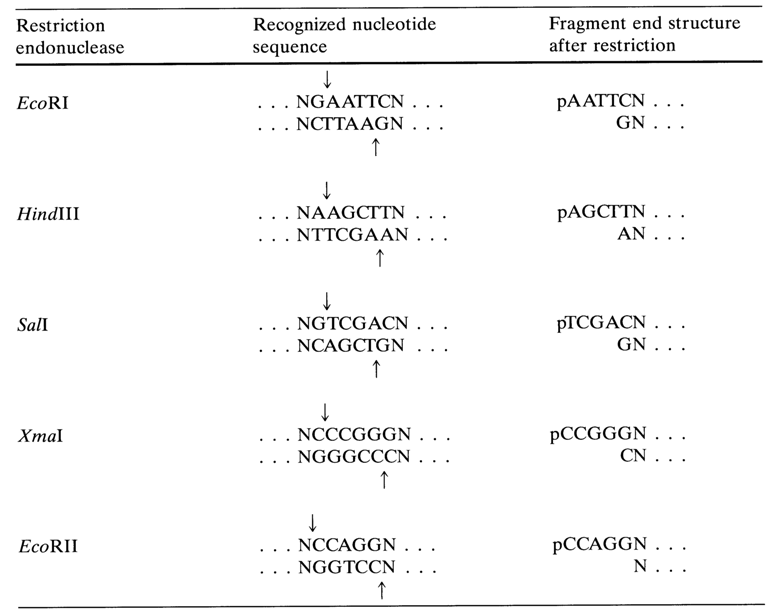
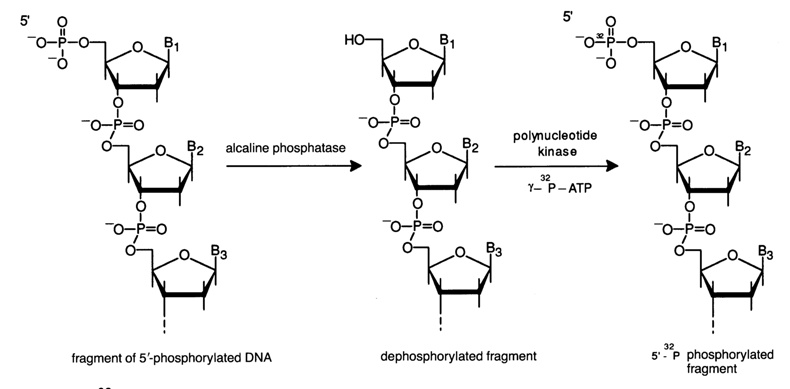
Fig. 6-5. 32P-labeling of the protruding 5' end of the restriction endonuclease-treated DNA fragment
An alternative method is known whereby the 5'-terminal phosphate of DNA is exchanged directly for a labeled phosphate with the aid of polynucleotide kinases and [
g- 32P]-ATP.Shown below (Fig. 6-6) is the standard procedure for preparing a set of duplex DNA fragments with one of the strands being labeled at the 5' end. The starting DNA, which may be a large restriction fragment or else a plasmid, phage or viral DNA, is exposed to a restriction endonuclease (E1). As a result of such restriction with subsequent phosphorylation short DNA duplexes labeled at both 5' ends are formed with the aid of a polynucleotide kinase, from which a labeled single-stranded DNA can be isolated after denaturation followed by gel electrophoresis. The mixture of labeled duplexes can also be treated with a second restriction endonuclease (E2). The result of this treatment is subjected to electrophoresis for isolating products with only one labeled strand.
The procedure is then repeated using the same restriction endonucleases (E1 and E2) but in the reverse order (Fig. 6-6, top). The illustrated hypothetical DNA fragment has three sites recognized by restriction endonuclease E1 and three sites recognized by E2. The restriction by means of a mixture of E1 and E2 gives seven duplexes (1 through 7 in Fig. 6-6). It can be seen that in this case only fragments 1 through 4, 6 and 7 are labeled at one strand. As regards fragment 5, it is either labeled at both strands or is not labeled at all (after treatment with E1 and then E2). Fragments 1 and 7 have only one strand labeled if they were treated with E2 at first and then with E1 (Fig. 6-6, top). Fragments 2, 3, 4 and 6 are labeled at the opposite strands, depending on the sequence of treatment with E1 and E2.
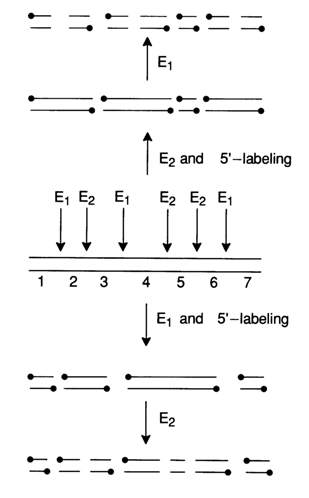
Fig. 6-6. Preparation of duplex DNAs 5'-end labeled at one of the strands only
Labeling the 3' Ends with the Aid of Terminal Transferase. Terminal transferase extracted from calf thymus polymerizes ribonucleotides in a template-independent reaction at the 3' ends of DNA strands. The function of substrates is performed by ribonucleoside 5'-triphosphates and, if the latter contain an et-labeled phosphate, such as [
a-32P] ATP, the 3' ends of DNA receive a polyribonucleotide labeled at the internucleotide phosphate.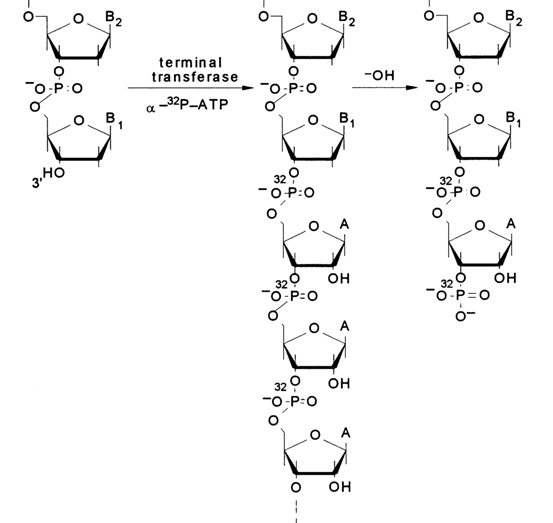
Alkaline treatment of such a mixed DNA-oligo-A-polymer gives a DNA with a "double" label at the 3' end: the 3'-terminal phosphate and the internucleotide one next to it are labeled. The mechanism of alkaline hydrolysis (the hydrolysis involves all internucleotide phosphate groups adjacent to the 2'-hydroxyl groups; i.e., only in the riboadenyl links) is represented schematically below:
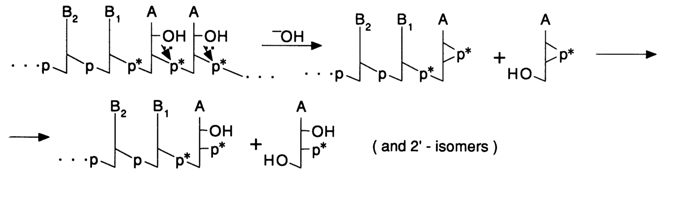
The first step of the alkaline treatment includes nucleophilic substitution at the internucleotide phosphorus with the participation of a 2'-hydroxyl. The resulting cyclic phosphates are then easily hydrolysed to a mixture of 2'- and 3'-phosphates. This process results in incorporation of two 32P-labeled phosphate groups into the 3' end of the DNA. 3' end labeling may be used in both single- and double-stranded DNAs. If pyrimidine nucleoside 5'-triphosphates are used as [
a-32P]-labeled nucleotides, alkaline treatment may be replaced by pyrimidyl RNase (the hydrolysis mechanism remaining the same).An alternative way to label the 3' end is extending the latter into duplex DNA with a protruding 5' end with the aid of an E. coli or phage T4 DNA polymerase. Here is a schematic representation of such a reaction in which the restriction fragment HindIII is extended:
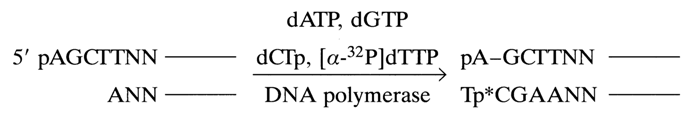
By using only dTTP containing an a.-labeled phosphate, one can incorporate the label into the 3'-terminal nucleotide of DNA.
An important step in extracting DNA from gel is its elution ensuring a high yield. Moreover, it is important to avoid degradation of DNA in the process. The extraction method must ensure that DNA is highly concentrated and free of the electrophoretic buffer, enzyme inhibitors and low-molecular weight polyacrylamide fragments. The best way is diffusion of DNA from the degraded gel in a salt solution with subsequent filtration and ethanol-assisted precipitation of the DNA from the filtrate. This is how DNA fragments varying in length from tens to thousands of nucleotides are extracted from polyacrylamide gel.
6.4.4 Polyacrylamide Gel Electrophoresis: High-Resolution System for Separating Oligo(poly)nucleotides According to Chain Length
The third basic principle of the sequencing method is separation of oligo- and polynucleotide chains according to size by way of gel electrophoresis. All labeled fragments resulting from the chemical degradation of DNA have one end in common and another end varying in length. Each fragment in the array contains all the nucleotides present in the preceding smaller fragment plus one more at the variable end. At pH > 7, two adjacent fragments differ by a small increment of charge and mass, one of the four mononucleotides. During PAGE, the larger fragment moves more slowly than the smaller one because it enters into greater noncovalent interaction with the gel matrix.
Currently used techniques permit separation of such DNA strands differing by a single nucleotide at lengths ranging from several to hundreds of nucleotides. Since these oligo(poly)nucleotides differing in length by a mere nucleotide appear on the X-ray film as bands, resolution is determined by two parameters: thickness of the bands and the distance between their centers. One must make sure that the concentration of the sample applied onto the gel is sufficiently high for the gel to be uniform, for diffusion to be weak, and for scattering of the radioactive emission to which the film is exposed to be modest. Then the bands on the film will be thin. The center-to-center distance between adjacent bands is determined by the "retarding" action of the gel matrix. Use is commonly made of thin 8 % polyacrylamide gels containing 50 % urea at pH 8.3 (8.3 M). Thin sequencing gels have important advantages: electrophoresis on such gels can be rather fast (at high voltage), and because scattering during autoradiography is reduced, the bands come out sharper on the film. Usually, within a single run of four standard reaction mixtures (G, G+A, C+T, C) anywhere between 100 to 150 nucleotides can be "read" in 8 % polyacrylamide gel. 20 % polyacrylamide gel is more suitable for "reading" the first 30 nucleotides from the labeled end.
For the purposes of autoradiography, the gel on the top of a glass plate is covered with thin plastic wrap and the plate is placed on X-ray film inside a light-tight holder. To slow down the "smearing out" of the bands during exposure, such holders are stacked in a freezer.
6.4.5 Direct Reading of Nucleotide Sequence from Autoradiogram
The great success of the chemical and Sanger's enzymatic sequencing techniques stems from the fact that the nucleotide sequence determined within a single analysis (an average of 300 nucleotides, the exact amount depending on the resolution of the gel electrophoresis) can be read directly from the autoradiogram. It would not be an exaggeration to say that the contribution of these techniques to molecular biology is more important than that of any other method or theory proposed since the discovery of the secondary DNA structure by Watson and Crick. Let us now see how a sequence of nucleotides is read. A typical autoradiogram of a sequencing gel is represented in Figure 6-7.
The autoradiogram shows vertical ladders of horizontal bands. Band at the bottom correspond to cleavage products near the labeled end or, in other words, short oligonucleotides. The upper bands correspond to cleavages progressively more distant from the labeled end. The spacing between the bands decreases with increasing length of the oligo(poly)nucleotides. The dark band interrupting the vertical row corresponds to the full-length DNA fragment not cleaved in all four reaction mixtures.

Fig. 6-7. Autoradiogram of a sequencing gel used in analysis of DNA 32P-Iabeled at the 5' end and its interpretation. The four vertical ladders correspond to reaction mixtures separated in 8 % polyaerylamide gel after limited cleavage at guanines (G), guanines and adenines (G+A), cytosines and thymines (C+T) and cytosines (C), using reactions 1-4 (Table 6-3).
In order to read the sequence of nucleotides (bases in the sugar-phosphate backbone of the polynucleotide) from these bands, let us look first at the bottom portion of the autoradiogram. The bands in the very bottom correspond to the short labeled DNA fragments. So we must move now from the bottom most band up, along the G+A and C+T ladders and interpret each band.
Note that these two ladders together contain all bands arising from partial cleavage of end-labeled DNA. If a band occurs on the G+A ladder and falls under G further to the left, it is a result of cleavage at guanine. If the G ladder is empty, then the band under G+A must result from cleavage at adenine. Similarly, a band on the C+T ladder to the right of the center line corresponds to cleavage at a pyrimidine base, whereas the presence or absence of a band under C suggests that the cleavage has taken place at cytosine or thymine, respectively. Thus, going from one band to another from the bottom upward allows one to immediately write down the sequence of nucleotides from the 5' end (which is labeled) of the DNA. The pattern on the film must be continuous, and the bands on the ladders must be sharply resolved and consistent with one DNA sequence. However, some bands tend to be doubled up, blurred, too light, or even missing. As a result, reading the DNA sequence becomes impossible. Such aberrations normally stem from improperly conducted chemical (insufficient specificity of modification reactions or incomplete P-elimination) and enzymatic reactions described earlier. Other reasons may be the presence of spurious fragments contaminating the main DNA fragment, DNA nicks, and microheterogeneous labeled DNA ends. A detailed description of the causes leading to all possible aberrations and ways to eliminate them can be found listed in a review by A. Maxam and W Gilbert (see references).
6.4.6 Double-Stranded DNA Sequencing Strategy
There are two main approaches to DNA sequencing. The first approach is used when it becomes necessary to determine the primary structure of a particular functionally significant DNA segment (a promoter, replication origin, structural gene, etc). It boils down to separation of the segment in question, after treatment of the sample, by means of a restriction enzyme and preparative PAGE. In this way, hundreds of picomoles of the DNA of interest can be obtained, which corresponds to about 1 mg DNA containing about 5000 pairs of bases. After electrophoresis, the gel is exposed to UV light against a fluorescent background. When this is done, the DNA bands appear dark. Once the desired band has been identified, the DNA fragment is extracted and precipitated with the aid of alcohol. The strands of such a fragment are most commonly separated by way of denaturation at 900C with subsequent electrophoreses and sequencing after 32P-labeling of the ends. It is still not clear what makes the complementary strands go apart during electrophoresis. In all likelihood, an important role is played by the configuration of each strand, which is dependent on the primary structure. The possibility to separate the strands permits one to clarify the primary DNA structure at the site of secondary restriction and to confirm the sequence when the second DNA strand is decoded. To separate strands up to 500 (and sometimes even more) nucleotides in length, use is made of 5 % PAGE. This technique is employed routinely when the DNA portion to be decoded is small and precisely mapped, as well as for separating cloned fragments of eukaryotic chromosomes from their bacterial vectors.
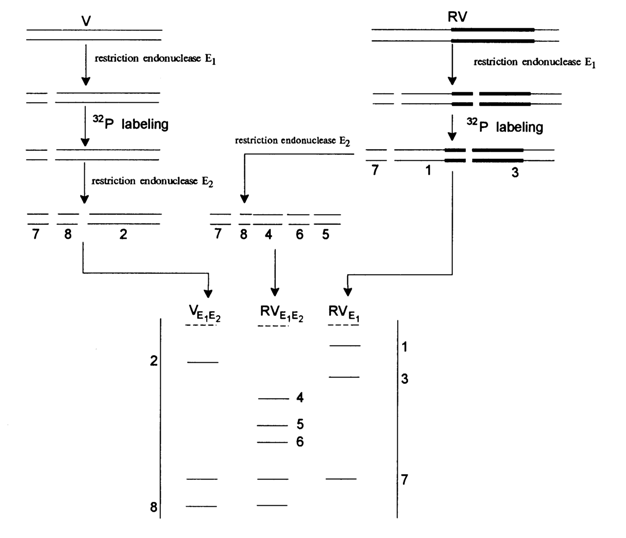
The second approach is used when it is necessary to sequence plasmid, phage or viral DNA. In this case, one can do without a detailed restriction map and a stockpile of isolated DNA fragments. At first, DNA is cleaved into fragments that are easy to label at the 5' or 3' ends (this purpose will best be served by restriction endonucleases which leave 5' ends extended). Then, the 5' or 3' ends of all these fragments are labeled at once. The subsequent analysis may proceed along two paths: (1) denaturation for strand separation and (2) digestion with a second restriction enzyme (see Figs. 6-4 and 6-6), followed by the separation of fragments labeled only at a single strand. If the scheme of Figure 6-6 makes use of the same two restrictases in reverse order, the result is the same set of fragments labeled at their opposite strand. Figure 6-8 illustrates the procedure of preparing fragments for sequencing of a cloned gene. It shows separation of fragments from a recombinant DNA (RV), based on the second path.
All fragments in the right ladder (RV) are labeled at both strands (because no treatment with a second enzyme was used), which is why they cannot be sequenced. All fragments in the left ladder (VE1, E2) lack inserts of the cloned gene under investigation. Only the bands (4, 5 and 6) in the central ladder (RVE1, E2) are fragments with a single label, include gene inserts and can be sequenced. Complete sequencing of both strands of a particular restriction
fragment is sometimes difficult by the above method. There are several reasons for this: the length of the strands may exceed the resolution of the sequencing genes; the separation of the strands may not be complete; difficulties may arise with determination of the terminal nucleotides. In such cases, the complementary strand is sequenced from the labeled 3' end, rather than the 5' end, or vice versa. One can also obtain fragments through digestion with another restriction enzyme and use newly emerging inner ends for sequencing. Such a method of sequencing over the entire length and along both strands is illustrated below.
The top portion of the scheme shows a DNA sequence containing sites of recognition by restriction endonucleases AluI, HaeIII, and a third enzyme. The sequence of this double-stranded DNA can be decoded as follows: treatment with restriction endonuclease AluI gives rise to a fragment that can be 32P-labeled at both 5' ends with the aid of polynucleotide kinase or at both 3' ends with the aid of terminal transferase. Then, restriction endonuclease HaeIII is used for asymmetric cleavage of the AluI-fragment, followed by sequencing of the resulting fragments from the labeled 5' (1 and 2) or 3' ends (3 and 4). It is also possible to cleave the strands of the AluI fragment and sequence the intact ones beginning from the labeled 5' (5 and 6) or 3' ends

(7 and 8). Moreover, the starting DNA may first be treated with restrictase HaeIII, labelled at the 5' or 3' ends, then treated with a third restriction enzyme cleaving the strand from both sides, and sequence in opposite directions from the HaeIII site, bypassing the AluI sites (9-12). All odd-numbered sequences validate completely or partially the even-numbered complementary sequence and vice versa. The results of all independent sequencing procedures are then represented by arrows pointing to the left and to the right, each arrow indicating which strand has been sequenced and which restriction site has been used for the purpose.
In conclusion, here is an example showing the results of sequencing of plasmid pBR322 (Fig. 6-9).
The transverse bands on top correspond to the sites of DNA pBR322 cleavage with restriction endonucleases HinfI and AvaII, and the restriction maps
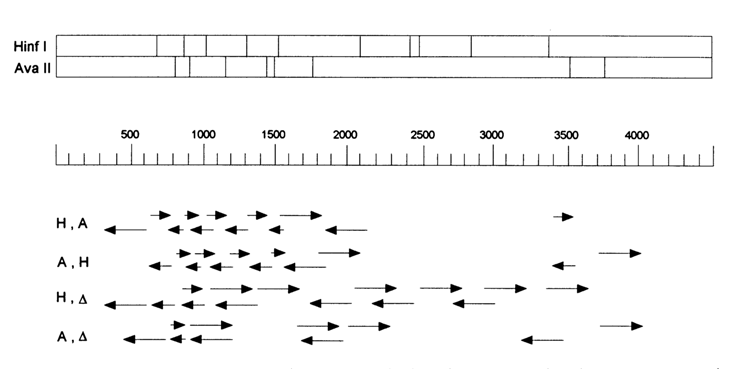
Fig. 6-9. Position of sequenced fragments of plasmid pBR322 labeled at one end (using two restriction endonucleases and polynucleotide kinase).
are arranged one above the other. The scale underneath represents the distances from the unique site of pBR322 cleavage with restriction endonuclease EcoRI in nucleotide pairs. The plasmid is 4362 base pairs long. Shown below the scale are rows of arrows. Each arrow begins from the 32P-labeled 5' end resulting from the cleavage with restriction endonucleases HinfI or AvaII, and its length equals that of the corresponding fragment, or 250 base pairs (resolution of gel electrophoresis) if the fragment is longer. The arrows mark the DNA sites next to the labeled ends, which can be decoded without any particular difficulty. The arrows in the row designated H,A correspond to the fragments labeled at one end and resulting from the following sequence of reactions: Hinfl
®kinase ® AvaII. The arrows in row A,H correspond to the fragments resulting from the reactions in reverse sequence: AvaII ® kinase ® HinfI. Both experiments have made it possible to sequence a total of 1700 base pairs, with both strands being read so as to verify and more unambiguously determine the sequence of each strand. The two bottom rows of arrows are designated H, A and A, A. They represent, respectively, the DNA fragments resulting from the following treatments: HinfI ® kinase ® melting and separation of strands (H,A) and AvaII ® kinase ® melting and separation of strands (A,A). Both experiments, carried out together with the previous two, have made it possible to determine the structure of the plasmid over 70 per cent (3000 b.p.) of its length.The vector most commonly used for cloning DNA fragments is plasmid pBR322, which contains six unique sites recognized by restriction endonucleases PstI, SalI, AvaI, HindIII, BamHI and EcoRI, occupying a small region of the lacZ gene. The DNAs inserted at these sites of plasmid pBR322 and then cloned can be sequenced by the Maxam-Gilbert method without complicated fragment treatment and separation procedures.
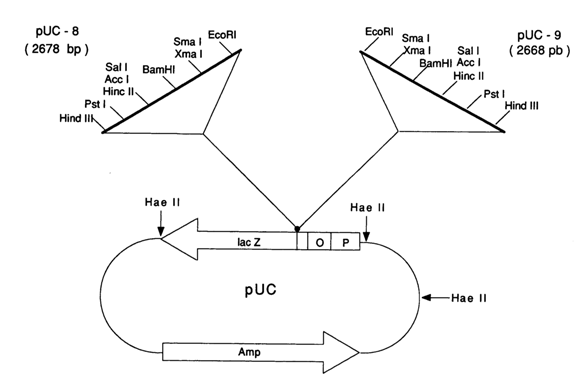
Fig. 6-10. Commercially available vectors pUC8 and pUC9
The company "Pharmacia" has been proposing vectors pUC for this purpose. They are essentially plasmids constructed on the basis of plasmid pBR322 and contain sites, over a portion of the lacZ gene, recognized by unique restriction endonucleases. Two such commercially available vectors (pUC8 and pUC9) are represented in Fig. 6-10.