![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
5.6 Sequence of Nucleotide Units
The order in which nucleotide units are arranged in a nucleic acid polymer chain is commonly known as nucleotide sequence. Determination of the nucleotide sequence in nucleic acids or their fragments is the primary task of nucleic acid chemistry and molecular biology. This is due to the fact that the sequence of monomer units in nucleic acids determines the content of their inherent genetic information, the ability to carry it, as well as the threedimensional structure and chemical properties of the polymer. Without knowing the primery structure of nucleic acids one cannot study the fine mechanisms underlying the functions of DNA and RNA. Knowledge of the primary structure of individual portions of nucleic acids makes it possible to construct synthetic models which serve as rather useful research tools in molecular biology, molecular genetics, enzymology, and many other sciences concerned with the functions of living organisms.
Systematic studies aimed at establishing the primary structure of nucleic acids began after methods for isolating some of them on an individual basis had been developed.
The first objects of such studies were transfer RNAs for which reliable isolation and purification techniques were elaborated in the early sixties. Since the molecular weight of tRNAs is relatively low, they provided a good basis for developing standard analytical procedures. At present, the primary structure of hundreds of individual tRNAs and some high-molecular weight RNAs is a matter of common knowledge.
The general approach currently used to determine the nucleotide sequence is basically the same as that proposed by Sanger to determine the amino acid sequence in proteins and known as the unit method. It consists of controlled fragmentation of biopolymer chains, separation of the resulting fragments (oligomers), and determination of the sequence of monomer units in these fragments. To reconstruct the original structure of the polymer, the analysis based on the above-described scheme should be repeated at least once, the second fragmentation involving other parts of the chain so that two sets of fragments have partially identical monomer unit sequences. Such an approach to reconstruction (establishment of the primary structure) can be schematically represented as follows:
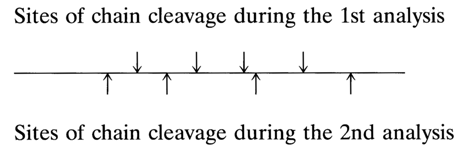
Comparison of the two sets of oligomers reveals overlapping sequences, which allows the fragments to be properly arranged one with respect to another. This is what is known as the overlapping unit method.
In the case of DNAs whose molecular weight is usually very high, the first analysis also involves the overlapping unit method but each unit is characterized by its molecular weight. It is this characteristic that permits the mutual arrangement of the units in the molecule to be determined using the above scheme (so-called mapping).
Until recently, the most commonly used technique for controlled cleavage of internucleotide bonds, as part of the unit method of analysing the primary nucleic acid structure, has been enzymatic hydrolysis or digestion.
In the mid seventies, radically new approaches were developed for determining the nucleotide sequence in nucleic acids, different from the unit method and allowing the rapid and reliable sequencing of high-molecular weight compounds. These methods, currently applicable to DNA and RNA and widely used for nucleic acid sequencing, will be discussed at greater length in Chapter 6.
The present chapter will deal with sequencing only in the context of oligonucleotides which are still valuable as effective means for determining the structure of synthetic oligonucleotides.
In what follows, we shall first of all discuss the general aspects of determining the primary structure of oligonucleotides, including the specific action of nucleases - enzymes breaking the internucleotide bonds in RNA and DNA.
5.6.1 Internucleotide Bond-Breaking Enzymes (Nucleases)
To determine the nucleotide sequence in oligo(poly)nucleotides and nucleic acids use is made of various enzymes breaking certain intermonomer bonds.
We shall first discuss enzymes of nucleic acid metabolism, which catalyze hydrolysis of polynucleotides and nucleis acids. Such enzymes are known as nucleases and classed with phosphodiesterases because they break internucleotide phosphodiester bonds. Used for structural studies in addition to phosphodiesterases, are also phosphomonoesterases which catalyze cleavage of the phosphomonoester bond with release of the phosphate from oligo(poly)nucleotides having a terminal phosphate group. The substrates of phosphomonoesterases are, as a rule, products of nucleic acid hydrolysis by nucleases.
The classification of nucleases is usually based on three characteristics. The first, fundamental characteristic is the substrate specificity of the enzyme that is, the ability of nuclease to hydrolyse RNA, DNA, or both. Hence such names as ribonuclease (abbreviated as RNase), deoxyribonuclease (DNase) and nonspecific nucleases whose abridged form, in the case of phosphodiesterases, is PDE plus the source from which it is isolated. The second characteristic is the way in which the polymer is digested by the enzyme or, in other words, its ability to catalyze hydrolysis of the polymer chain endolytically that is, at a particular site inside the chain, or exolytically - that is, beginning from the end of the chain and breaking step by step the terminal internucleotide bonds. This is how the terms endonucleases and exonucleases came into being. Finally, the third characteristic is the type of cleavage of a particular internucleotide linkage which may be hydrolysed in two ways depending on its structure. For instance, cleavage of the internucleotide linkage in the shown fragments is possible both between the 3'-hydroxyl and phosphorus atom, giving a 5'-phosphate group and freeing the 3'-hydroxyl, and between the phosphorus and 5'-hydroxyl, giving terminal 3'-phosphate and 5'-hydroxyl groups:
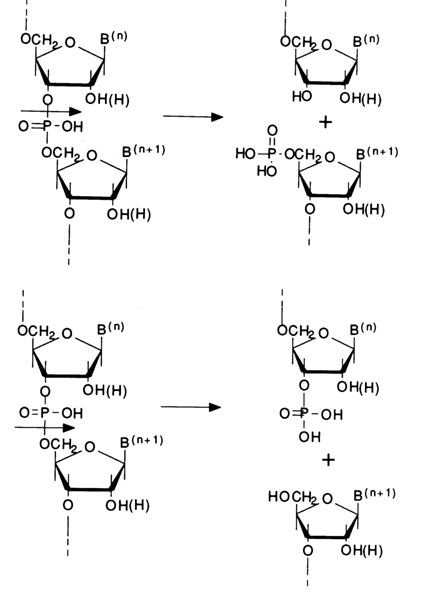
This characteristic is usually ignored in the name of the enzyme, the assumption being that the mechanism of phosphodiester bond cleavage by the enzyme, whose name takes into account the first and/or second characteristics, is known. We shall now consider the mechanism of action of the nucleases most widely employed in studying the structure of oligo(poly)nucleotides and nucleic acids.
5.6.1.1 Ribonucleases
Pyrimidyl Rihonuclease. The most fully studied enzyme in this category is pyrimidyl nuclease (abbreviated as RNase A). It was discovered in 1920 in the pancreas and was crystallized by Kunitz in 1940 (J. Gen. Physiol. 24, 15). The molecular weight of the enzyme is 13,700; it is stable in a broad pH range and very stable when heated in weakly acid solutions. The activity is maximum at pH 7.7 (650C). The enzyme has no effect on DNA. Pyrimidyl RNase exhibits high specificity of endolytic action: it catalyses hydrolysis of any internucleotide 3'-5' bonds formed by the 3'-hydroxyl of the pyrimidine nucleotide. The nucleotide acting as donor of the 5'-hydroxyl may be either a pyrimidine or purine one. What makes the action of this enzyme special is that it splits RNA only over single-stranded portions without affecting the double-stranded ones.
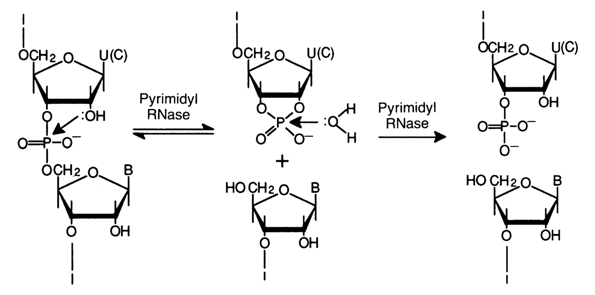
The cleavage of internucleotide linkages proceeds in two steps. The
first step is transphosphorylation (intramolecular nucleophilic substitution involving the
2'-hydroxyl) to yield an oligonucleotide terminating in a 2',3'-cyclic phosphate group of
the pyrimidine nucleotide![]() . The second step is hydrolysis of the
cyclic phosphate group (intermolecular nucleophilic substitution involving a water
molecule) to yield an oligonucleotide with the terminal 3'-phosphate group of the
pyrimidine nucleotide. The general scheme of RNA cleavage by pyrimidyl ribonuclease is
given below:
. The second step is hydrolysis of the
cyclic phosphate group (intermolecular nucleophilic substitution involving a water
molecule) to yield an oligonucleotide with the terminal 3'-phosphate group of the
pyrimidine nucleotide. The general scheme of RNA cleavage by pyrimidyl ribonuclease is
given below:
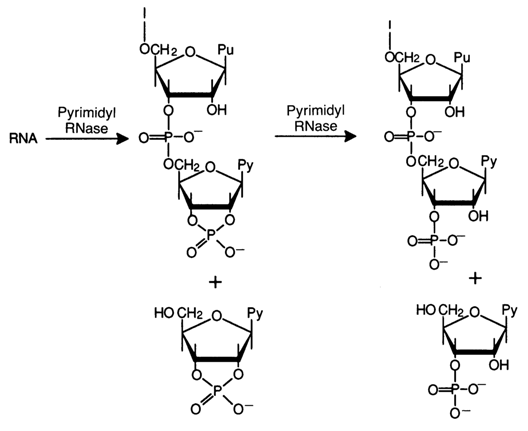
The products of short-term treatment of RNA with pyrimidyl ribonuclease are invariably pyrimidine ribonucleoside 2',3'-cyclic phosphates and purine oligoribonucleotides with pyrimidine nucleoside 2',3'-cyclic phosphate at the 3' end of the oligonucleotide chain.
Thus, pyrimidyl ribonuclease is essentially a highly specific phosphodiesterase catalysing the degradation of diphosphates in which one alcohol group must be represented by the 3'-hydroxyl of pyrimidine ribonucleoside. The other alcohol group does not have to be a nucleoside. As demonstrated in experiments on model substrates, pyrimidyl RNase also hydrolyses pyrimidine ribonucleoside 3'-alkylphosphates.
Synthetic pyrimidine ribonucleoside 2',3'-cyclic phosphates are also easily hydrolysed in the presence of pyrimidyl RNase.
To illustrate the action of the enzyme on polyribonucleotides, here is a scheme showing the cleavage of an 18-membered fragment forming part of a tRNA:
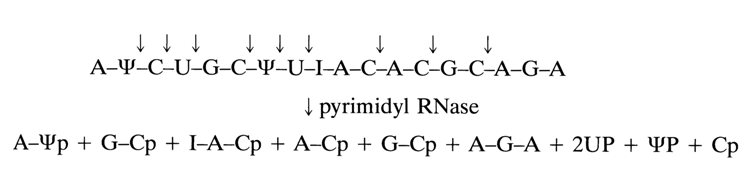
As a result of such digestion, four dinucleotides are formed along with two trinucleotides and three pyrimidine nueleotides. A distinctive structural feature of the hydrolysates is the presence of pyrimidine nucleotides at their 3' end. Adenosine turned out to be 3'-terminal only in one of the trinucleotides, which immediately indicates that this oligonucleotide occupies the 3' end of the polynucleotide chain. As can be inferred from the above scheme, the internucleotide linkage involving the pseudouridine 3'-phosphate group is broken just as similar linkages formed by uridine and cytidine 3'-phosphates. It should also be noted that the specificity of pyrimidyl RNase with respect to pyrimidine nucleotides is not absolute. The enzyme hydrolyses the internucleotide bonds formed by the adenosine 3'-phosphate group, albeit at a much slower rate (by about two orders of magnitude).
Guanyl Ribonuclease. The second enzyme of this group, isolated from socalled takadiastase which is an extract from the mold Aspergillus oryzae is known as ribonuclease T, (RNase Tl). Similar enzymes have been isolated from actinomyces and other sources. The specificity of ribonuelease Tl is narrower than that of pyrimidyl RNase, because it breaks only one type of internucleotide linkage, namely, bonds formed by the 3'-phosphate group of guanosine (and its derivatives) and the 5'-hydroxyl of any neighboring nucleotide. By virtue of the specificity of its action, RNase T, is often referred to as guanyl ribonuclease. The enzyme cleaves only single-stranded portions of RNA.
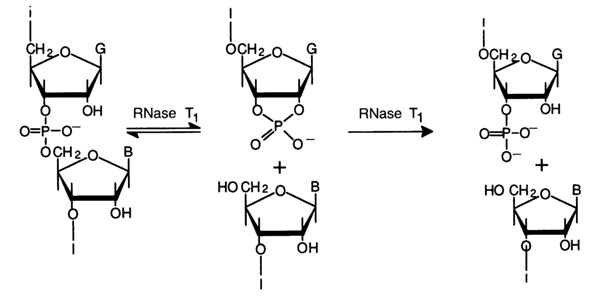
Just as in the case of pyrimidyl RNase, hydrolysis proceeds in two steps with intermediate formation of guanosine 2',3'-cyclic phosphate or an oligonucleotide with a 3'-terminal guanosine 2',3'-cyclic phosphate. The first step is reversible; the cyclic phosphate hydrolysis rate is much slower than that of transphosphorylation. And as with pyrimidine RNase, the specificity of ribonuclease T1 is not absolute. The enzyme hydrolyses the internucleotide bonds formed by the inosine 3'-phosphates methylated at the pyrimidine ring, guanosine 3'-phosphates, and also xanthosine 3'-phosphates (in the latter case, hydrolysis is much slower). Now follows a scheme showing how ribonuclease T, splits the same 18-membered polynucleotide which was used to illustrate the action of pyrimidyl RNase:
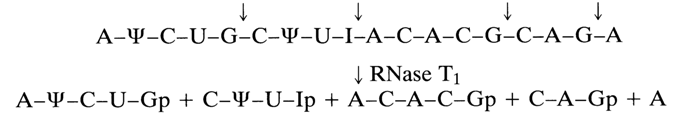
Comparison of the mechanisms of action of pyrimidyl RNase and RNase T1 shows that the oligonucleotide units resulting from treatment with the second enzyme are much larger. A characteristic feature of RNase T1 hydrolysates is the presence of guanosine 3'- or minor inosine 3'-phosphates at the 3' end of the oligonucleotides. Studies into hydrolysis of model compounds with RNase T1 have shown that this enzyme has many potential uses in nucleotide chemistry. Guanyl RNase is one of the enzymes that have played a decisive role in structural investigations of RNA.
5.6.1.2 Deoxyribonucleases
The first enzymes of this type were isolated from the pancreas (pancreatic DNase or DNase 1) and spleen (DNase 11). Both have turned out to be endonucleases.
Pancreatic Deoxyribonuelease (DNase 1). This enzyme has been found to lack pronounced specificity toward bases as it breaks some internal linkages. At cleavage sites, the phosphate group remains linked to the 5'-hydroxyl:
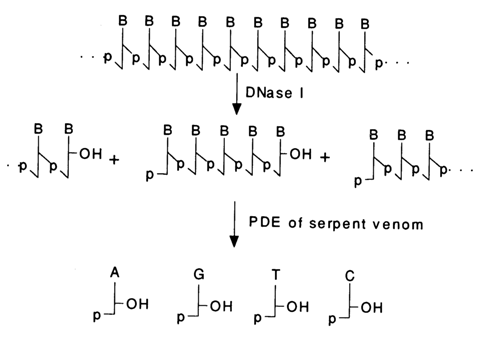
The resulting oligonucleotides are good substrates for snake venom PDE (see below), which becomes useful in preparative hydrolysis of DNA to deoxyribonucleoside 5'-phosphates.
Spleen deoxyribonuclease (DNase 11). This enzyme, just as DNase 1, is not very specific with respect to bases and cleaves internucleotide bonds in DNA in such a manner that the phosphate group remains at the 3'-hydroxyl:
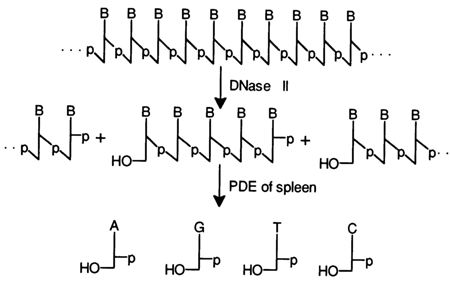
The products of DNA hydrolysis in the presence of DNase 11 serve as substrates for spleen PDE (see below). Such DNA hydrolysis to nucleotides is used in structural studies but seldom applied to preparative isolation of the corresponding deoxyribonucleotides.
In addition to the DNases just described, there are many other deoxyribonucleases (both endo- and exonucleases). These enzymes have yet to be completely studied and sometimes are not readily available. Restriction enzymes and endonuclease IV should be mentioned among the DNases already employed in structural studies.
Endonuclease IV. This enzyme was isolated for the first time in 1969 from E. coli infected with phage T4. The enzyme most effectively catalyses the cleavage of the internucleotide linkage formed by the 3'-hydroxyl of deoxythymidine and 5'-phosphate group of the deoxycytidylic acid and acts only on single-stranded DNA. The result is oligonucleotides one of which contains the 3'-terminal deoxythymidine and the other has a deoxycytidine 5'-phosphate at the 5' end:
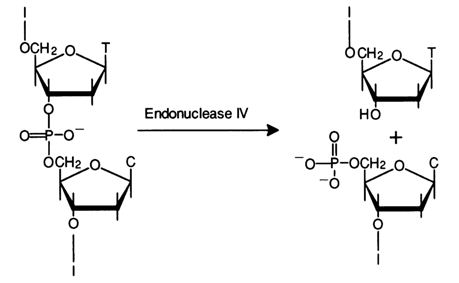
As regards other internucleotide linkages, endonuclease IV cleaves also the dGpdC bond, but at a much slower rate.
5.6.1.3 Non-specific Nucleases
Phosphomonoesterases (PME). The enzymes belonging to this group are sometimes referred to as phosphatases. They cleave phosphomonoester bonds in nucleotides or in ribo- and deoxyribooligo(poly)nucleotides with a 5'- or 3'-terminal phosphate group.
Because they break any phosphomonoester bonds, phosphomonoesterases are known as non-specific. This category of PME includes alkaline phosphatase from E. coli, acid phosphatase from the prostate, and alkaline phosphatase from wheat. In their presence, nucleotides are degraded to nucleosides, while oligonucleotides with terminal phosphate groups are converted into those with free 3'- and 5'-hydroxyls; a molecule of inorganic phosphate is removed in the course of the reaction:
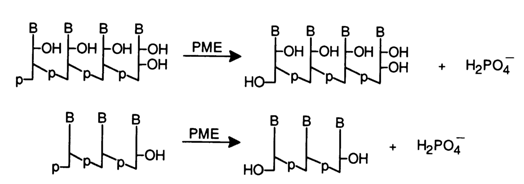
Specific phosphomonoesterases are treated elsewhere. In addition to these PMEs hydrolysing only nucleotides mention should be made of 3'-nucleotidase found in E. coli. This enzyme removes 3'-terminal phosphate groups in RNA and DNA.
Snake Venom Phosphodiesterase. The enzyme is isolated from the venom of cobras, vipers, and so on. It cleaves RNA and DNA step by step beginning from the 3'end with removal of nucleoside 5'-phosphates.
When hydrolysis is conducted under conditions when not all internucleotide linkages have time to be cleaved (limited amount of the enzyme, low temperature), a statistical set of molecules is formed including both starting molecules and those shortened from the 3' end by one, two, three and more nucleotide units.
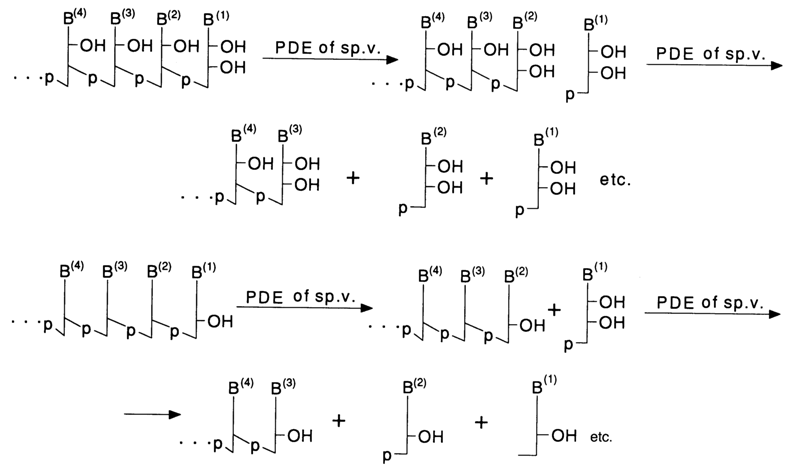
The presence of a phosphate group at the 3' end of an oligo(poly)nucleotide in the substrate inhibits the enzyme. In such cases, snake venom PDE is used after treating the oligo(poly)nucleotide with phosphomonoesterase. This enzyme is employed for preparative separation of deoxyribonucleoside and ribonucleoside 5'-phosphates, respectively, from DNA and RNA.
Spleen Phosphodiesterase. This enzyme catalyses cleavage of oligo(poly)nucleotides from 5' end of the polymer chain. Each catalytic event breaks only the terminal internucleotide linkage with removal of nucleoside 3'-phosphate. When hydrolysis is not complete, a statistical set of molecules shortened from the 5' end by one, two, three and more nucleotide units is formed, just as in the case of snake venom PDE.
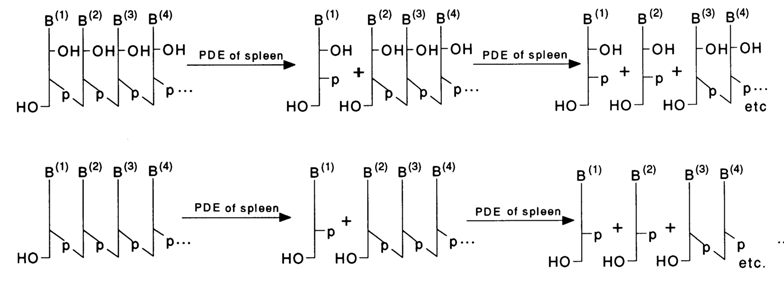
The enzyme does not cleave polynucleotides containing a phosphate group at the 5' end.