![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
5.6.2 Methods for Determining the Nucleotide Sequence in Oligonucleotides
As has already been mentioned, determination of the nucleotide sequence in nucleic acids is a rather difficult task and must be accomplished in stages: at first, the polynucleotide chain must be split into units or, to be more precise, oligonucleotides, and then one must determine the nucleotide sequence in each oligonucleotide chain. Analysis of chains consisting of three or four monomer units is not difficult, but as the chain becomes longer, the difficulties increase progressively. Should this be the case, a lot depends on the nature of individual nucleotide units and their mutual arrangement in the oligomer chain.
Oligoribonucleotides. The procedure of analysing the structure of oligoribonucleotides comprises several steps, the nucleotide composition being determined first, then the terminal fragments and, finally, the sequence of monomer units.
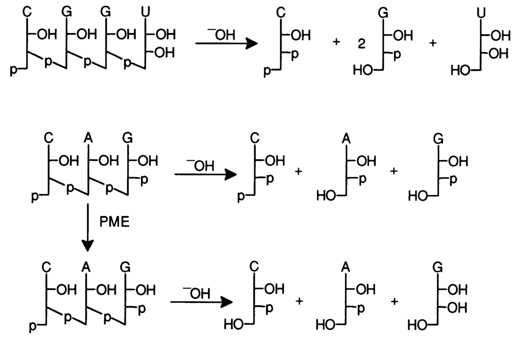
The easiest way to determine the nucleotide composition of
oligoribonucleotides is through alkaline hydrolysis with subsequent identification of the
corresponding ribonucleoside 3'(2)-phosphates forming as a result; for example: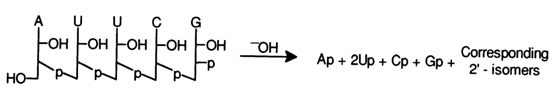
Exonucleases, such as snake venom PDE, can be used for the same purpose:
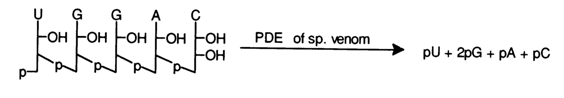
The determination of the terminal fragments of the molecules (so-called terminal analysis) may also involve chemical and enzymatic hydrolysis. Alkaline hydrolysis makes it possible to determine the nature of the terminal fragments within a single run, provided the terminal phosphate group is linked to the 5' hydroxyl (the scheme does not show the corresponding 2'-isomeric nucleotide products):
In addition to the usual products of alkaline hydrolysis, ribonucleoside 3'(2')phosphates, the hydrolysate in this case also contains 3',5'-diphosphonucleoside (5'-terminal fragment) and a free nucleoside (3' -terminal fragment), which can be separated and identified by paper chromatography and electrophoresis.
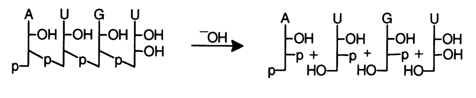
The nature of the terminal nucleotides may also be determined by way of complete hydrolysis in the presence of snake venom and spleen PDE, if the terminal phosphate groups are removed in advance. For instance:
It can be seen that as a result of treatment with snake venom PDE the nucleoside is formed from the 5'-terminal fragment of the molecule, and if spleen PDE is used, the source of the nucleoside is the 3'-terminal fragment. Thus, by using spleen PDE one can get the same information as after alkaline treatment, which is why alkaline hydrolysis is preferred for determining the 3'terminal groups in oligoribonucleotides - alone or in combination with enzymatic hydrolysis.
Terminal analysis of polynucleotides may be used to determine the length of the polymer chain (it is the numerical ratio between terminal fragments and the rest of nucleotides that is determined).
If it is necessary to find out whether the polymer has phosphate groups at its ends, it is treated with PME. Appearance of a phosphate in the hydrolysate indicates that terminal phosphate groups are present.
In determining the nucleotide sequence in short oligonucleotides it is sometimes quite sufficient to resort to alkaline hydrolysis and/or digestion with snake venom PDE. This is illustrated by the above example and the examples that follow (also formed are the corresponding 2-isomeric nucleotides which are not shown):
Analysis of longer oligoribonucleotides can be based on two approaches: consecutive removal of nucleotides from one of the ends of the oligonucleotide or its cleavage at the internucleotide linkages using enzymes of different specificity with subsequent establishment of the order in which monomers alternate in the units and reconstruction of the starting oligonucleotide (method of overlapping units).
The consecutive removal of monomer units by a chemical method was for the first time performed in Todd's laboratory. The procedure begins from the 3' end after periodate oxidation of the 2',3'-cis-diol group in the oligoribonucleotide. The resulting dialdehyde is selectively cleaved under mild conditions in a
b-elimination process (the emerging carbonyl group facilitates detachment of the proton at C") to give an oligonucleotide shortened by one monomer, a dialdehyde fragment, and a base.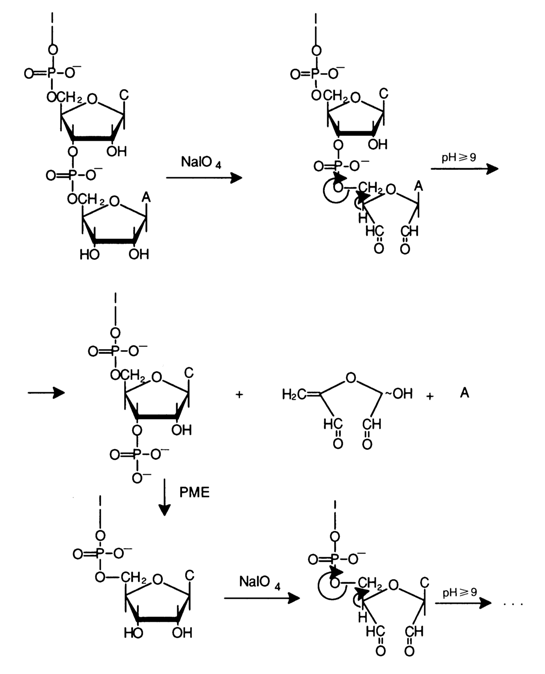
Since reliable methods for analysing bases are available, one can easily determine the nucleotide with the 3'-terminal group which was removed.
Enzymatic dephosphorylation in the presence of phosphomonoesterase yields an oligonucleotide shortened by one monomer, again containing a 2',3'-diol group prone to oxidation, which can undergo the same sequence of transformations. A prerequisite for successful application of this method is completeness of each of the three consecutive reactions (oxidation,
b-elimination, and dephosphorylation) as well as absence of nuclease traces in the phosphomonoesterase preparation used. If these conditions are not met. by-products rapidly accumulate in the reaction mixture and analysis becomes complicated.The above procedure makes it possible to determine the nucleotide sequence in oligoribonucleotides rather reliably. It has also been used to determine the 3'-terminal sequence in tRNA. A case in point is establishment of the 3'-terminal sequence in phenylalanine tRNA from E. coli having a length of 19 nucleotides.
The consecutive removal of nucleotides from the ends of an oligonucleotide can also be achieved in the presence of exonucleases. Short-term treatment of an oligoribonucleotide with snake venom PDE gives all possible oligonucleotides - a statistical set (as opposed to the above-mentioned step-by-step shortening of the chain) of molecules undergoing no changes and shortened by one, two, three and more monomer units. For example:
pApUpUpCpCpG
pApUpUpCpC
pApUpUpC
pApUpU
pApU
Nucleoside 5'-phosphate
The resulting oligonucleotides are separated by ion-exchange chromatography and 3'-terminal nucleotides are determined in each of them. It can be easily seen that the enumeration of 3'-terminal nucleotides in the unchanged oligonucleotide and then in that shortened by one monomer unit and so on all the way to the dinucleotide (the two nucleotides are enumerated in the latter) will represent the primary structure of the oligomer under analysis, beginning from the 3' end. This approach is widely used at present to determine the nucleotide sequence in oligodeoxyribonucleotides.
The enzymatic method of determining the nucleotide sequence, involving removal of the terminal nucleotides with the aid of PDE, has its limitations similarly to the chemical method.
For instance, when long oligonucleotides are treated with an enzyme,
individual cleavage of internucleotide linkages are observed in the middle of the chain
because of the presence of trace amounts of other enzymes. As a result, attempts to
determine sequences of eight to ten monomer units in this fashion have seldom been
successful. Another approach most widely used to determine the nucleotide sequence in
oligoribonucleotides involves cleavage at internucleotide linkages with subsequent
separation of short oligonucleotides, establishment of their structure, and reconstruction
of the starting molecule. Here are some simple examples illustrating the method under
discussion (in both cases, the 3'-terminal nucleotide is known ![]()
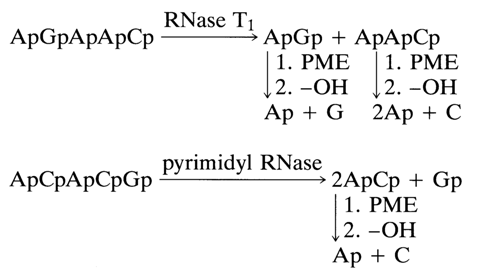
A partial hydrolysis method has been developed for analysis of oligonucleotides containing polypyrimidine sequences whose structure cannot be determined by complete hydrolysis with pyrimidyl RNase. During such hydrolysis under mild conditions, some internucleotide linkages normally cleaved during complete hydrolysis may remain intact, which makes it possible to isolate oligopyrimidine units. Subsequent analysis of partial degradation products allows one to easily determine their structure and then the structure of the starting oligonucleotide. The following example may illustrate such an approach. The following abridged formula has been written for the heptanucleotide forming a part of valine tRNA (A. A. Baev, T V Venkstern et al.) after determination of the general nucleotide composition and terminal nucleotides (remember that nucleoside symbols separated by commas in parentheses stand for an unknown sequence):
A (A,UY,C,C)Gp
After partial hydrolysis of this heptanucleotide in the presence of pyrimidyl RNase two trinucleotides were separated, their structure being determined rather easily:
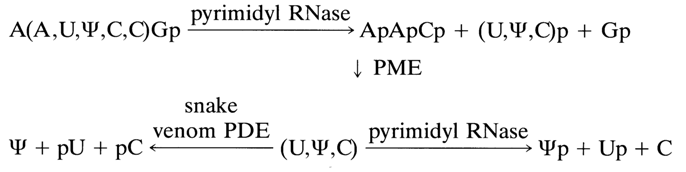
These results give unambiguously the following nucleotide sequence in the starting heptanucleotide:
ApApCp'YpUpCpGp
Another example (also from the same source) shows how one can gradually approach the true structure in the case of the hexanucleotide (A,UW,C,C)Gp for which complete enzymatic hydrolysis does not provide an unambiguous answer. The data necessary for determining the structure of this hexanucleotide are listed in Table 5-2.
The above examples show that determination of the oligoribonucleotide structure is a more or less difficult problem which must be solved in a particular way for each individual case.
Oligodeoxyribonucleotides. When the nucleotide sequence is determined in oligodeoxyribonucleotides, the composition and terminal fragments are analyzed first, just as in the case of oligoribonucleotides, and only then the entire sequence.

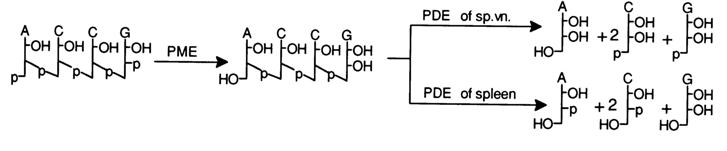
The nucleotide sequence may be determined by resorting to acid hydrolysis under vigorous conditions (72 % perchloric acid, 1000 C, or 85 % formic acid, 1750 C), during which a mixture of pyrimidine and purines bases is formed as a result of cleavage of N-glycosidic bonds. In general, however, the nucleotide composition is analyzed together with the terminal fragments by way of complete enzymatic hydrolysis with exonucleases - snake venom and spleen PDE. Again, as in the case of oligoribonucleotides, the terminal phosphate is removed with the aid of PME, then treated with PDE under conditions when all internucleotide linkages are broken, for instance:
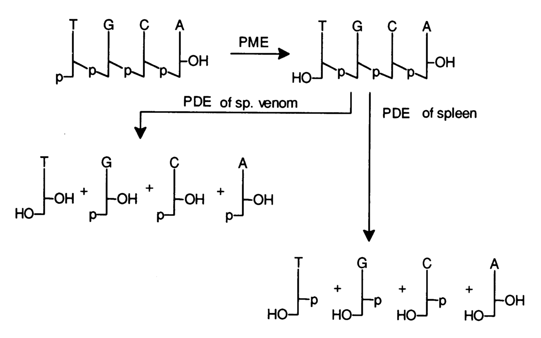
The composition of the nucleosides found in both hydrolysates reveals the
nature of the 5'- and 3'-terminal units of the oligodeoxyribonucleotide, while the ratio
between the amounts of the emerging nucleoside and nucleotides ![]() gives an indication of the chain length and
composition of the starting oligodeoxyribonucleotide.
gives an indication of the chain length and
composition of the starting oligodeoxyribonucleotide.
The main difficulty in determining the primary structure of
oligodeoxyribonucleotides stems from the fact that until recently no enzymes have been
found to be as specific as pyrimidyl and guanyl RNases with respect to
oligo(poly)nucleotides ![]() .
.
Only one approach has been elaborated for determining the primary structure of oligodeoxyribonucleotides, namely, partial hydrolysis with phosphodiesterases. Just as with oligoribonucleotides, the problem boils down to separation of the oligomer mixture resulting from digestion with the number of nucleotide units gradually going down to two. The analysis of terminal nucleotides in each emerging oligomer makes it possible to determine the order in which nucleotides alternate in the starting molecule.
It should be pointed out that the most difficult task is to find the conditions when the hydrolysate would contain the entire set of nucleotides from the unreacted starting one to the dinucleotide. The oligodeoxyribonucleotide under analysis is first treated with PME, then either with snake venom or spleen PDE. In the former case, the set of oligodeoxyribonucleotides is formed as a consequence of removal of the monomeric 5'-phosphates from the 3' end of the oligomer chain, and in the latter, as a result of removal of the monomeric 3'-phosphates from the 5' end. Given below are the sets of oligonucleotides resulting from incomplete degradation of a dodecadeoxyribonucleotide, aided with snake venom and spleen PDE:

In the beginning of analysis one may obtain any one of the above sets
from the starting oligonucleotide. Then, each individual component (except for the
mononucleotides) is isolated to determine which nucleotide is terminal. If a set of
oligonucleotides is obtained using snake venom PDE, the 3'-terminal fragments are
determined in each by separating them in the form of nucleosides under the action of
spleen PDE. For example: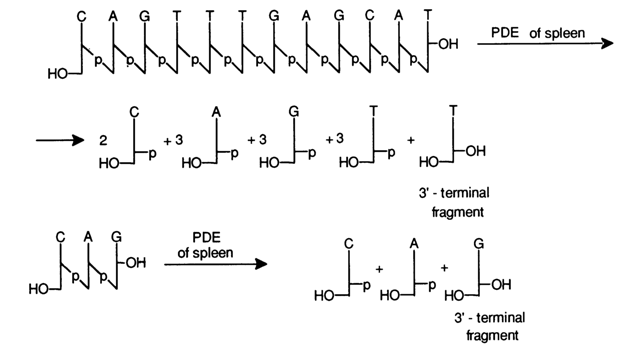
And if a set of oligonucleotides is derived through hydrolysis of the starting oligomer in the presence of spleen PDE, then the 5'-terminal fragments are determined (in the form of nucleosides) in each isolated oligonucleotide using snake venom PDE. For example:
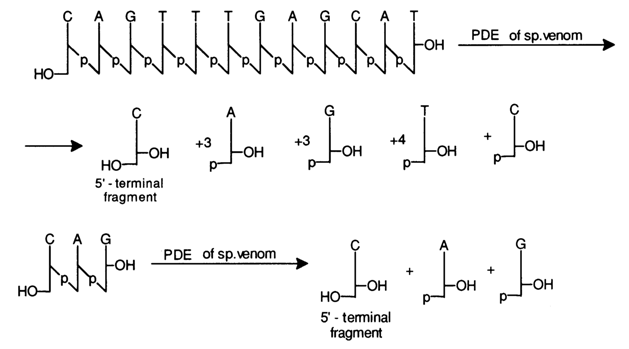
The coincidence of the results obtained in the two ways described above is indicative of their reliability.
However, the need to isolate each oligonucleotide resulting from hydrolysis of the sample under investigation and subject each of them to terminal analysis renders the method too cumbersome, especially when we are dealing with longer oligomers.
With this in view, Sanger recently developed a method, sometimes called the "wandering-spot" method, which allows the nucleotide sequence in oligodeoxyribonucleotides to be determined without isolation of each individual oligonucleotide resulting from partial digestion of the sample under analysis. It includes preparation of so-called nucleotide maps (see below), which are essentially the results of two-dimensional separation of oligonucleotides obtained through enzymatic hydrolysis (first, in the presence of PME for removing the terminal phosphate groups, then, as has already been described, snake venom or spleen PDE). A 32P-labeled terminal phosphate group is introduced into the oligonucleotides under analysis so that one could work with minimal amounts of the substances. The 5'-terminal hydroxyls undergo phosphorylation. The reaction is conducted in the presence of the enzyme polynucleotide kinase and
g- 32P-ATP: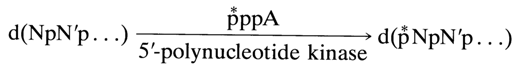
Nucleotides containing labeled phosphate (indicated * in the above scheme) are easily detectable when sensitive X-ray films are used (autoradiography).
Two-dimensional separation of a mixture of 5'-phosphorylated oligodeoxyribonucleotides obtained in the above-described fashion proceeds in two steps: first, the mixture is subjected to electrophoresis on a narrow strip of cellulose acetate at pH 3.5 in one direction; then, the result is placed on a DEAE-cellulose plate so that it serves as the starting line in thin-layer chromatography with an aqueous solution of the oligoribonucleotide mixture produced during alkaline hydrolysis of RNA being used as the eluent (this process is known as "homochromatography"; oligonucleotides without labeled phosphorus do not impede the determination). The method offers a clear separation of labeled oligonucleotides differing in length just by a single unit.
In order to understand the results of two-dimensional separation or, in other words, be able to "read the nucleotide map" one must know the extent to which the mobility of the oligonucleotide changes in the course of electrophoresis and chromatography as a result of removal of a mononucleotide unit. The mobility of oligonucleotides during electrophoresis depends both on the net negative charge of the molecule and on its size: the greater the charge, the greater the mobility, the latter dropping as the molecular weight increases. Therefore, a substantial decrease in charge even with simultaneously diminishing molecular weight will result in a particle with reduced electrophoretic mobility, whereas a small decrease in charge with a simultaneous decrease in molecular weight will give a particle with increased mobility. The difference in total charges of the nucleotides stems from the fact that cytosine, adenine, and guanine exhibit different basicity - that is, protonation capacity and, consequently, ability to carry a positive charge. At pH 3.5, the phosphate groups in nucleotides carry only a negative charge, and the bases differ in protonation capacity. This is why electrophoresis should be conducted at pH 3.5. In this case, the net charges of deoxycytidine, deoxyadenosine, deoxyguanosine and deoxythymidine phosphates approach -0.15, -0.45, -0.9 and -1, respectively.
Consider two extreme cases. If thymidine phosphate is removed from an oligodeoxyribonucleotide or, in other words, the molecule loses a negative charge, and in spite of the marked decrease in molecular weight, whatever is left of the oligonucleotide becomes electrophoretically less mobile; and if cytidine phosphate with a net charge of only -0.15 is removed and the molecular weight is still considerable, the electrophoretic mobility of the oligonucleotide with reduced molecular weight and volume, but not charge, turns out to be greater, as compared to the starting oligodeoxyribonucleotide. The mobility of oligonucleotides during chromatography under pre-set conditions always increases inversely with the length of the oligonucleotide chain. Thus, the oligonucleotide becomes more mobile as it loses mononucleotide units.
Let us now see how nucleotide maps will be affected by the above regularities. The removal of deoxycytidine and deoxythymidine phosphates will change the nucleotide map as shown below on the top.
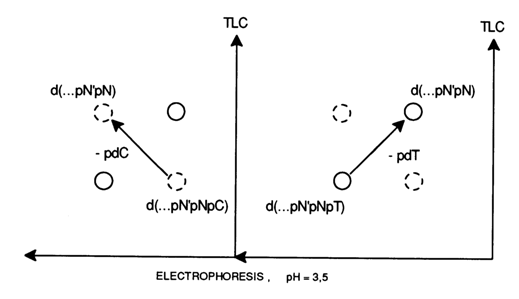
It can be seen that the two-dimensional movement of the oligonucleotide shortened by one unit is given, depending on the nature of the removed unit, by the angle formed with the perpendicular to the starting line of the unshortened nucleotide.
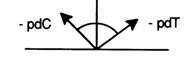
The more chromatographically mobile of every two adjacent spots on the
two-dimensional map is a product of removal of the terminal unit from the less mobile
precursor. If it is also more mobile electrophoretically, the removed nucleotide is
deoxycytidylic acid, and if it is less mobile, then we are dealing with deoxythymidylic
acid. Given below by way of example is a two-dimensional map produced during analysis of
the undecadeoxyribonucleotide d(C5T6) sequence after incomplete
digestion with snake venom PDE: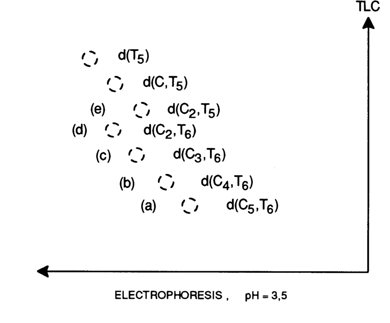
Spot (a) corresponds to the starting oligodeoxyribonucleotide d(C5T6). The next spot (b), which is more mobile chromatographically, is a product of removal of a nucleotide from the 3' end of the starting oligonucleotide. Since compound (b) is also more mobile electrophoretically, pdC is the detached unit. Thus, it may be assumed that the 3'-terminal unit in d(C5T6) is pdC. The two following spots (c) and (d) seem to be products of sequential removal of two more deoxycytidine 5'-phosphate groups; that is, the formula of the starting oligonucleotide can be written as follows:
d[(2C,6T)pCpCpC
Compound (e) is electrophoretically less mobile than (d), yet, as can be inferred from chromatographic mobility, it is formed from compound (d) as a result of removal of a nucleotide. The implication is that compound (e) is formed from (d) as a consequence of removal of deoxythymidylic acid; that is, the formula of the oligonucleotide can be written as
d[(2C,5T)pTpCpCpC
Complete analysis of the map gives the 3'-terminal sequence of the oligonucleotide as
d(... pCpCpTpCpCpC);
that is, the structure of the starting compound is
d(pTpTpTpTdpTpCpCpTpCpCpC).
This is how the nucleotide sequence in many oligodeoxyribonucleotides containing pyrimidine nucleotides has been determined.
Two-dimensional (chromatography-electrophoresis) maps of oligonucleotides also containing purine deoxyribonucleotides reveal the same pattern: removal of all nucleotides (both pyrimidine and purine ones) leads to higher chromatographic mobility of the fragment, yet during electrophoresis its mobility remains virtually the same after removal of deoxyadenylic acid whose loss brings about a decrease in the net charge (by -0.5) along with a tangible molecular weight reduction. Removal of pdG results in lower electrophoretic mobility of the fragment (the net charge of the molecule is reduced by -0.9) but to a lesser extent than in the case of pdT removal.
The change in mobility of an oligodeoxyribonucleotide during two-dimensional chromatography-electrophoresis as a result of removal of pyrimidine and purine nucleotides can be shown schematically as follows:
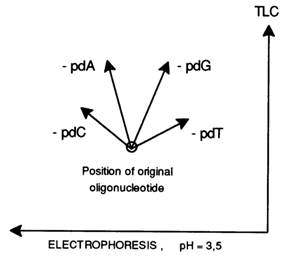
Note that removal of purine nucleotides increases the chromatographic mobility of the remaining oligonucleotide to a much greater extent than that of pyrimidine ones.
Evidently, this phenomenon cannot be explained by the fact that purine nucleotides have a greater molecular weight because the difference in the letter is relatively small (pdC 227, pdT 242, pdA 251, pdG 267); a more likely reason is the pronounced adsorption capacity of purine bases (on DEAE-cellulose in the case under consideration).
Shown below is a nucleotide map of d(C2,T4,A2,G4) - a dodecanucleotide containing all four deoxyribonucleotides. After partial digestion with spleen PDE, all eleven compounds became so arranged that it was possible to determine the initial nucleotide sequence:
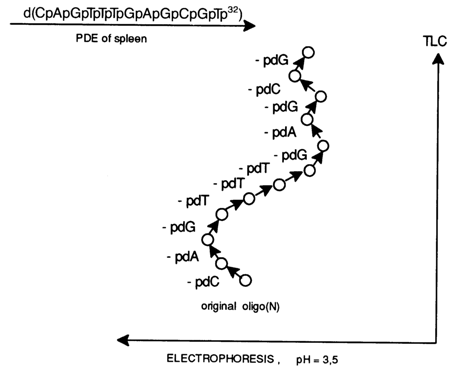
This approach was used to determine the nucleotide sequence of many oligodeoxyribonucleotides isolated from hydrolysates of phage and other DNAs.
Among the drawbacks of the method is its inability to obviate the difficulties that arise while selecting the right conditions for partial digestion of the oligonucleotide under analysis with phosphodiesterases, which would make it possible to obtain a complete set of oligonucleotides - from the intact starting one to that consisting of two units. Moreover, the method may involve errors stemming from small changes in mobility during analysis of rather long oligonucleotides. The best results were obtained while studying oligonucleotides with no more than ten monomer units.
At present, the Maxam-Gilbert method is also used to determine the primary structure of oligonucleotides.