![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
2 Properties of Nucleosides
2.1 Heterocyclic Bases
An unsubstituted pyrimidine is an aromatic system in which, just as in
pyridine, the carbons are deficient in electrons due to electronegativity of the nitrogens
![]() . There are two factors contributing to
this, namely, induction and mesomeric effects. The former is especially pronounced at
positions next to the nitrogens - that is, 2, 4 and 6. Position 5 is electrondeficient but
to a lesser degree.
. There are two factors contributing to
this, namely, induction and mesomeric effects. The former is especially pronounced at
positions next to the nitrogens - that is, 2, 4 and 6. Position 5 is electrondeficient but
to a lesser degree.
The mesomeric effect may manifest itself only in one direction, with electrons being shifted toward the nitrogen, otherwise unviable six-membered cyclic structures with a cumulative system of multiple or triple bonding would be formed.
The electron-attracting mesomeric effect of the nitrogen atoms N1 and N3
may be represented by the following resonance structures: ![]()
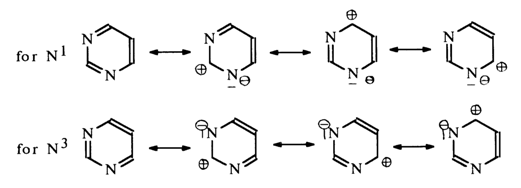
The above structures show that the mesomeric effect of the nitrogen atoms also leads to electron deficiency in the carbons at positions 2, 4 and 6. Consequently, the aromatic ring in pyrimidine is stable toward electrophilic reagents even to a greater extent than in pyridine. The only site vulnerable to attack by an electrophilic reagent is position 5. Therefore, the latter is often referred to as aromatic. It is also natural that positions 2, 4 and 6 must be reactive in reactions with nucleophilic reagents. These principles also apply to unsubstituted pyrimidine, which is confirmed by experimental evidence, albeit rather scant because this compound is extremely difficult to isolate. Various derivatives of pyrimidine, whose reactions are more complex yet are governed by the same basic principles, have been studied much more comprehensively.
In considering the purine ring (containing an imidazole nucleus in addition to the pyrimidine one) it is appropriate to draw an analogy between the imidazole and pyrrole nuclei.
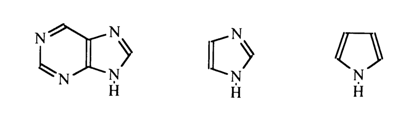
The lone-pair electrons of nitrogen in pyrrole are involved in formation of the aromatic sextet - that is, they become incorporated into the nucleus. The distribution of electron density in the pyrrole nucleus can be represented by the following resonance structures:
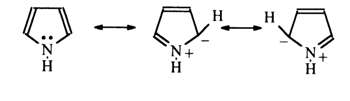
The electronegativity of nitrogen in this case manifests itself only in polarization of the N-H bond, which leads to increased mobility of the proton associated with the nitrogen atom. As a result, electrophilic substitution in the case of pyrrole is facilitated primarily in the a-positions. Thus, the pyrimidine ring in the purine nucleus is marked by surplus of p-electrons. It is expected that positions 2 and 6 in purine are chemically similar to positions 2 and 4 in the pyrimidine ring. Position 8 may be compared to the a-position in pyrrole. It should be taken into account, however, that the p -electron clouds of both monocyclic systems in purines overlap and that the true electron density distribution in each ring may change as a consequence of electron donation by the imidazole ring to the pyrimidine one.
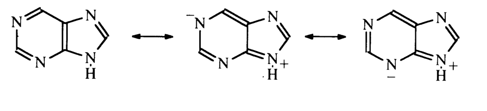
In the presence of electron-donor groups in the pyrimidine moiety, the electron density in the imidazole ring will increase.
The pyrimidine and purine rings, just as those of pyridine and pyrrole, are two-dimensional and all of the hydrogen atoms linked with them lie in the same plane, with the electron sextet in each nucleus, just as in benzene, forming regions of increased electron density above and below the ring plane.
Such are the qualitative concepts ![]() usually invoked when discussing the
chemical behavior of pyrimidines and purines.
usually invoked when discussing the
chemical behavior of pyrimidines and purines.
The ability of heterocyclic bases to undergo tautomeric interconversions markedly influences the three-dimensional structure of nucleic acids as well as the reactivity of the bases themselves, both at the monomer (as part of nucleosides and nucleotides) and polymer levels.
Since nucleic acids contain hydroxy and amino derivatives of purine and pyrimidine, these compounds may display, similarly to hydroxy and amino derivatives of pyridine, two types of tautomerism: lactim-lactam and enamine-ketimine:
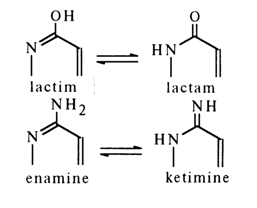
As regards hydroxy derivatives, if they are in the lactam form, the lone-pair electrons of the "amide" nitrogen may lead to formation of a new aromatic system. No rupture of conjugation at the carbonyl carbon takes place because
C=C - C - C=C systems are characterized by
both direct and cross-conjugation.
Another argument in favor of the lactam form is the greater affinity to the proton of the nitrogen atom, as compared to the oxygen atom.
In the case of amino derivatives, it is generally more difficult to chose between the enamine and ketimine forms, although the above assumptions favor the enamine form. Thus, for example, the lactam-enamine form of tautomerism will be more preferable for cytosine and guanine:
3)
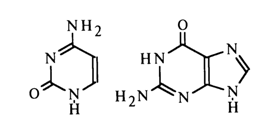
In view of the fact that substituents are present at N1 of pyrimidine bases and N9 of purine bases in nucleic acids and their monomer units, the possibilities of tautomeric transitions are limited.
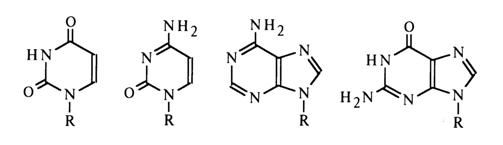
Experimental proof supporting the existence of particular tautomeric forms of pyrimidines and purines in appropriate nucleosides (in aqueous solutions) was provided by UV, IR and NMR spectroscopy. The bases of nucleic acids were compared with substituted pyrimidines and purines in which a particular tautomeric form is possible. In the case of uridine, for instance, N3-methyluridine and other methylated uracils were used for comparison. At neutral pH values, uracil in uridine may exist in the following three forms:
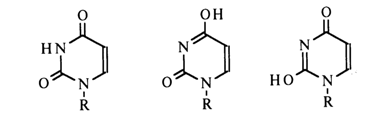
The IR spectra of uridine and N3-methyluridine incapable of enolyzation turned out to be virtually identical, thereby suggesting that uridine in aqueous solutions exists in a diketo form. This conclusion was also confirmed by the difference between the IR spectra of uridine and its 4-ethoxy derivative which seemed to possess one of the enolic forms. Moreover, it was established that the UV spectrum of uracil resembles more closely that of 1,3dimethyluracil (1) and differs markedly from that of 1-methyl-4-ethoxydihydropyrimid-2-one (2).
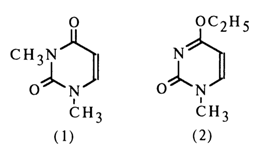
The NMR spectra of uridine and thymidine in nonaqueous solutions were found to feature a single resonance peak of the proton of the N-H group, which also supports the conclusions drawn from the UV and IR spectra to the effect that uridine and thymidine exist in a diketo form. According to the results of IR spectroscopy and X-ray structural analysis, the diketo form was also ascribed to 5,6-dihydrouridine (a minor nucleoside).
The similarity between the UV and IR spectra of cytidine (3), deoxycytidine (4) or N1-methylcytosine (5) and their 4-dimethylamino derivatives has led to the conclusion that cytosine nucleosides exist in a keto-amino form:
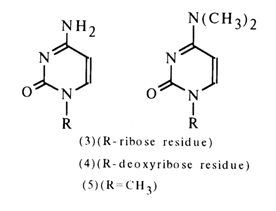
Final proof supporting this conclusion was provided by the finding that the NMR spectrum of cytidine contains a peak typically associated with an aromatic amino group and lacks the peak corresponding to the proton of the N-H group.
Theoretically, adenosine may exist both in imino and amino forms. The similarity between the IR spectra of 6-amino and 6-dimethylamino derivatives favors the latter.
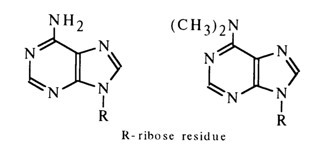
This conclusion is corroborated by the NMR spectra of adenosine and deoxyadenosine, having a characteristic double-proton peak which corresponds to the amino group, but no peaks corresponding to other protons associated with nitrogen.
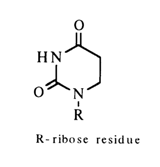
Comparison of the IR spectra of guanosine (6), 1,9-dimethyl-guanine (7) and 9-b-D-ribofuranosyl-2-amino-6-methoxypurine (8) in D2O reveals close similarity between the structures of the first two compounds and their difference from the structure of the third compound lacking the carbonyl group (R being ribose residue).
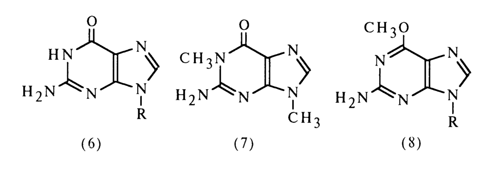
When the NMR spectra of guanosine and 1,9-dimethylguanine were compared, it was found that the single-proton peak in the spectrum of guanosine corresponds to the proton bound to N1. The fact that the singlet double-proton peak is not split means that both protons are bound to the same atom which therefore can only be the nitrogen of the 2-amino group. All this is the evidence in support of the keto-amino structure of guanosine.
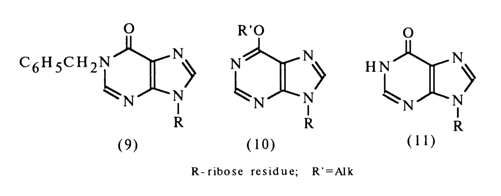
Comparison of the IR spectra of inosine and its derivatives with tautomeric forms (9) and (10) has shown that it has a keto structure (1l):
If one proceeds from the assumption that the electron density in unsubstituted nuclei of pyrimidine and purine is discontinuous, or split (see above), as well as from the data on the most preferable tautomeric forms of existence of the major pyrimidine and purine bases in nucleosides, one can draw conclusions about the distribution of electron density in the corresponding substituted heterocyclic systems:
Atoms donating two electrons to the common 7-electron system (nitrogen atoms of the pyrrole type, such as N1 in pyrimidine and N9 in purines, as well as those of exocyclic amino groups) must exhibit some electron deficiency (61). At the same time, atoms contributing a single electron to the aromatic system and having a pair of free electrons (endocyclic nitrogen atoms of the pyridine type: N3 of cytosine, N1 of adenine, and also N3 and N7 of adenine and guanine) must exhibit a partial negative charge (d-). Accordingly, it is these atoms that must be affected by electrophilic reagents in the first place.
Therefore, all of the carbon atoms, except for C5 in pyrimidines, must react with nucleophiles. The latter atom is more likely to interact with electrophiles. Such electron density distribution is consistent with calculation results. The reactivity of pyrimidine and purine bases in nucleosides and in nucleic acids also supports the above.
2.1.3 Reactions with Electrophilic Reagents
Substitution Reactions at Carbon Atoms. These reactions include substitution for the protons bound to the carbon atoms at position 5 in pyrimidine bases or position 8 in the purine ring. They lead to modification of the heterocycle in the nucleoside without breaking it. Halogenation is the most common of these reactions. Free halogens in a nonaqueous medium trigger direct substitution for the hydrogen (at C5) in pyrimidine nucleosides:
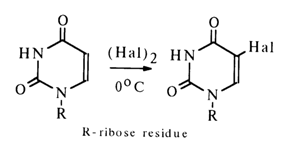
The reaction proceeds under mild conditions with a practically quantitative yield. This has been used to obtain 5-chloro and 5-bromo derivatives of uridine and cytidine. Iodination requires much more vigorous conditions - with heating and the presence of nitric acid which seems to be necessary to remove the hydrogen iodide reducing the iodo derivative produced during the reaction. When ICl is used as the iodinating agent, the reaction proceeds under much milder conditions. In the presence of water, addition at the double bond involving C5 and C6 takes place in the reaction mixture.
In the case of purine nucleosides, where the competing reaction of addition at the above-mentioned double bond is impossible, halogenation proceeds in both aqueous and nonaqueous solvents:
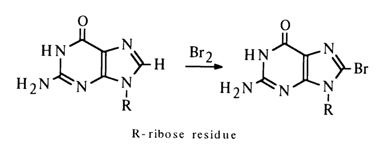
Guanosine lends itself more readily to halogenation than adenosine. 8-Halogenpurines are rather unstable compounds and are easily hydrolysed during halogenation with rupture of the imidazole ring.
Nitration of nucleosides requires much more vigorous conditions (higher temperature, nitrating mixture). Therefore, one must protect the hydroxy groups of the carbohydrate moiety in the nucleoside against oxidation.
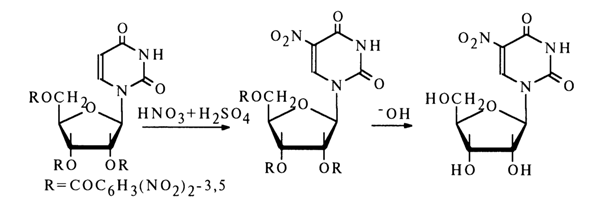
The corresponding 5-amino nucleosides can be obtained by reduction of the 5-nitro derivatives.
Heating of uridine together with formaldehyde in the presence of hydrochloric acid leads to its 5-hydroxymethylation.
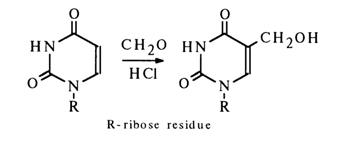
Under similar conditions, cytidine does not undergo hydroxymethylation (probably because of protonation of the amino group whose positive charge prevents electrophilic substitution).
5-Hydroxymethyluracil is easily formed when the base is hydroxymethylated in an alkaline medium.
Aminomethylation takes place in the presence of hydrochlorides of secondary amines and formaldehyde.
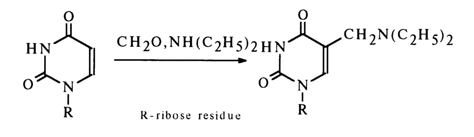
5-Hydroxymethyl and 5-aminomethyl derivatives can be converted into the corresponding 5-methylnucleosides through hydrogenation over a platinum or rhodium catalyst.
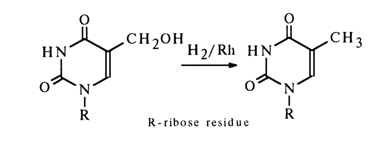
These reactions are widely used in nucleoside chemistry for their ability to easily produce 5-methyl and 5-hydroxymethyl derivatives as well as their analogues.
Substitution Reactions at Nitrogen Atoms. The most typical and widely used reactions of this type are those of alkylation with diazomethane, alkyl halides and alkyl sulfates. Other common reactions include those of addition at polarized C=C and C=N bonds and also those yielding N-oxides.
Methylation. The behavior of pyrimidine and purine derivatives during the methylation with diazomethane is largely dependent on the reaction conditions. In the absence of protic solvents the predominant process is substitution of a methyl group for the hydrogen atom exhibiting the most acidic properties - that is, substitution at N3 in uracil derivatives (this may be equated with the need for preliminary protonation of diazomethane) and at N1 in guanine derivatives.
The methylation can be represented by the following scheme:
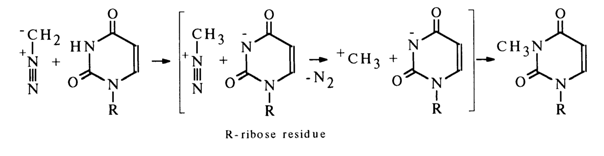
If other proton donors are also present in the reaction mixture, the methylation of the pyrimidine or purine ring proceeds at the site with the highest electron density because diazomethane becomes electrophilic in the course of the reaction without participation of the heterocyclic base.
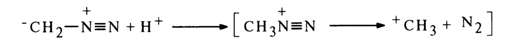
For instance, when a guanosine suspension in ether is treated with diazomethane, the reaction yields N1-methylguanosine, whereas the reaction in a water-ether medium gives N7-methylguanosine:
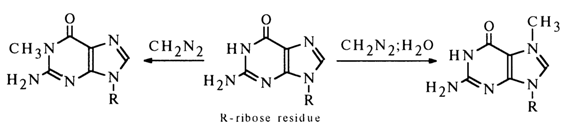
The methylation of uracil nucleosides at the heterocycle always involves only N3; cytosine nucleosides are methylated only in the presence of water, giving N3-substituents:
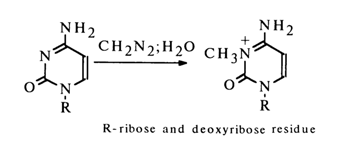
The propensity of nucleosides for interaction with diazomethane in water-ether solutions decreases as follows: guanosine ~ uridine > cytidine > adenosine.
In addition to diazomethane, methyl iodide and dimethyl sulfate are also used for the methylation of bases. The alkyl radical in the molecules of both substances is associated with electronacceptor groups which are substituted, when interacting with sufficiently strong nucleophiles, and act as departing groups. During the reaction, the alkyl (methyl) radical is transferred to the nucleophile and alkylation of the latter occurs. In this particular case, heterocyclic bases act as nucleophiles. When treated with these reagents in a neutral medium, guanosine, adenosine and cytidine undergo methylation, whereas uridine and thymidine do not enter into the reaction.
Guanosine is alkylated giving N7-methylguanosine; in the presence of potassium carbonate (K2C03), however, the main product of the reaction is N1-methylguanosine.
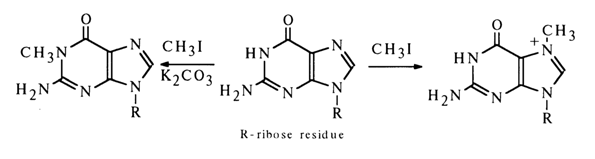
It is most likely that in the presence of K2C03 the -NH-CO group in the heterocyclic
ring of guanosine is partially converted into the anionic form -N=C-O- which
(just as most ambidentate ions) reacts with methyl iodide at the
most nucleophilic site or, in other words, at the nitrogen atom.
Methylation of adenosine proceeds primarily at N1, and that of cytidine, at N3.
The ease with which methylation by such agents as methyl iodide and dimethyl sulfate takes place changes in the following decreasing order: guanosine > adenosine > cytidine >> uridine.
Interaction with Reagents Having C=N and C=C Bonds. The heterocyclic moieties of nucleosides react readily with compounds having C=N and C=C bonds. The mechanisms of such reactions seem to boil down to nucleophilic addition at the polarized double bond.
Examples include reactions with carbodiimides and acrylonitrile. They usually involve heterocycles with -CO-NH- groups, primarily uridine:
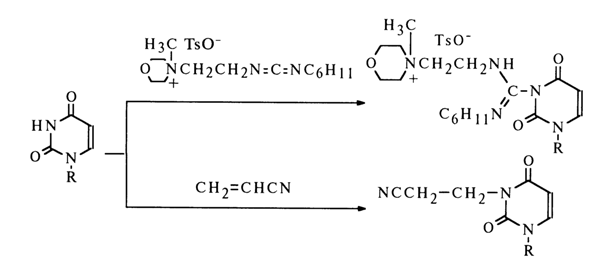
Other substances reacting with carbodiimide include thymidine and guanine nucleosides as well as such minor nucleosides as inosine and pseudouridine.
Oxidation with Peracids. Similarly to many nitrogen-containing heterocycles, heterocyclic bases in nucleosides are oxidized with peracids to yield N-oxides. For example, oxidation of adenosine in the presence of hydrogen peroxide and acetic acid gives N1-oxide:
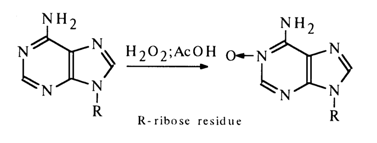
Cytidine is oxidized with arylcarboxylic peracids to yield N3-oxide:
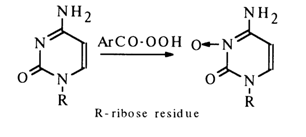
Isotopic Exchange of Hydrogen Atoms. The substitution of deuterium and tritium for hydrogen atoms in the purine and pyrimidine rings may proceed, depending on the reaction conditions, as electrophilic or protophilic exchange. The hydrogen atoms linked to nitrogen are exchanged most rapidly, while in the case of those bound to carbons the exchange is much slower. The exchange of the isotope back for hydrogen is also rapid.
Of particular interest for nucleoside studies are samples containing a more or less strongly bound hydrogen isotope and, in this connection, proton exchange reactions at the carbons of the heterocyclic base. If we take protons of low mobility, those bound to the carbon of the imidazole ring (C8) in purine bases are exchanged more easily than others, and the exchange rate increases with increasing pH of the medium and with temperature.
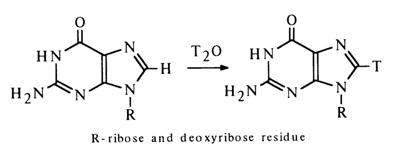
Nucleosides labeled in this fashion are employed in biological experiments that are usually conducted at neutral pH values and temperatures not exceeding 400C - that is, conditions ruling out the loss of the label. The exchange in pyrimidine derivatives proceeds under more vigorous conditions, the hydrogen at C5 being exchanged more easily than at C6.
2.1.4 Reactions with Nucleophilic Reagents
Reactions of heterocyclic bases with nucleophilic reagents bring about substitution of functional groups (amino groups as a rule) in the pyrimidine or purine nucleus. This is why they arouse a great deal of interest for modification of nucleic acids. The reactions with hydroxylamine, O-alkylhydroxylamines as well as hydrazine and its derivatives are the most common.
Depending on pH, hydroxylamine may react selectively with different bases of nucleic acids. At acid and neutral pH values, it reacts with cytosine:
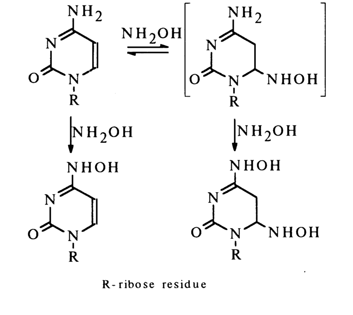
The fastest step of the reaction is the nucleophilic addition of hydroxylamine at the double bond involving C5 and C6. The subsequent nucleophilic substitution at C4 gives both di- and monohydroxyamino derivatives of cytosine. O-Alkylhydroxylamines react in a similar manner. The reaction rate reaches its maximum at pH 5-6, which indicates that cytidine enters into the reaction in a protonated form, while hydroxylamine (pKa 6.5) does so as a free base.
The reaction of hydroxylamine and O-methylhydroxylamine with adenosine is markedly slower (optimal pH value is 4 or 5). It yields exclusively products of substitution of a hydroxylamino or O-methylhydroxyamino group for the amino one.
The amino group in guanosine does not seem to be modified by hydroxylamine and its O-alkyl derivative. Only hydroxylamine reacts with uridine and only in an alkaline medium. This reaction leads to the rupture of the heterocycle. This is why it will not be considered here, although the reaction is extensively used for specific modification of nucleic acids at uracil units.
Another group of nucleophilic reagents is represented by hydrazine and its derivatives. The reactions involving them are conducted in an aqueous medium, their mechanism being heavily dependent on the pH of the latter. Nucleophilic substitution occurs only at pH 7 with only cytosine nucleoside entering into the reaction; the reaction products are mono- and disubstituted hydrazines:
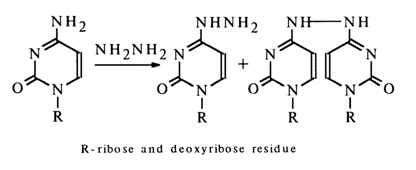
The reaction with monomethylhydrazine proceeds under similar conditions and yields a single product:
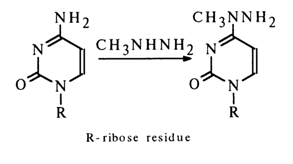
Purine nucleosides as well as uridine and thymidine do not react with hydrazine and its derivatives under the above conditions.
In an alkaline medium or nonaqueous solvents hydrazine breaks down uracil and cytosine derivatives. In contrast, it has virtually no effect on purine nucleosides at all.
Apart from the reactions discussed above, nucleophilic substitution for amino groups in heterocyclic bases proceeds in the presence of an alkali and amines. Cytosine nucleosides are the most reactive in this case.
It is known that C5, C6-dihydrocytidine undergoes deamination most easily in the presence of an alkali; this reaction seems to proceed as follows:
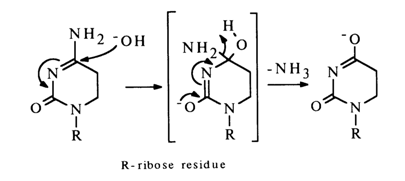
In the case of cytidine, the deamination seems to be preceded by hydration giving the corresponding dihydro derivative which is first deaminated as shown above, then undergoes dehydration yielding uridine.
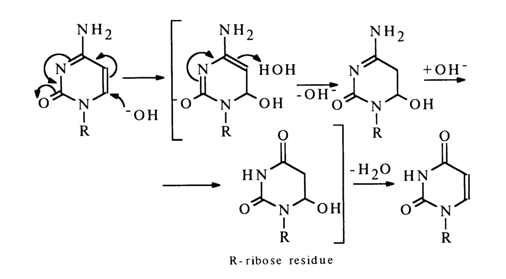
Under similar conditions, 5-methyldeoxycytidine reacts at a slower rate, as compared to deoxycytidine, which is quite consistent with the reaction mechanism. Attempts to make adenosine enter into this reaction are successful only under vigorous conditions.
Cytidine and deoxycytidine undergo reamination in aqueous solutions:
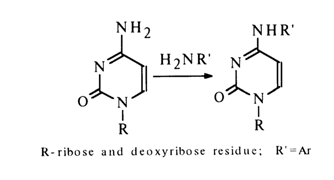
The reactions of amino group substitution in nucleosides, especially in the case of cytosine derivatives, are widely used in nucleic acid chemistry as a means for controlled modification of bases. Nucleophilic substitution reactions also find use in synthetic chemistry of nucleosides. The substitution of sulphur for the carbonyl oxygen (e.g., in uracil derivatives) also belongs to nucleophilic substitution reactions. For example:
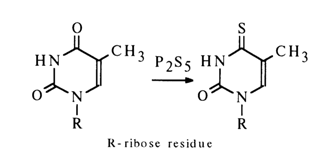
As a result of incorporation of thio groups into ribo- and deoxyribopyrimidine nucleosides, reactive intermediates are formed, from which both major natural nucleosides and their various analogues can be obtained. For instance:
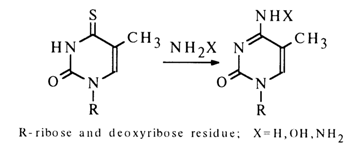
These and similar reactions of nucleophilic substitution have become quite popular in the modification of pyrimidine and purine bases which are subsequently subjected to ribosylation as part of synthesis of natural nucleosides and their analogues with 2-, 4- and 6-substituents.
These reactions are typical of pyrimidine derivatives with a reactive C5-C6 double bond. They have become instrumental as a means for imparting greater lability to the glycosidic bond in pyrimidine nucleosides when analyzing nucleoside structure as well as during the synthesis of various analogues of nucleosides as part of studies into the mechanisms of their action.
Halogenation of uracil and cytosine nucleosides in an aqueous medium has long been the most fully studied addition reaction. It proceeds in several steps and results, under certain conditions, in addition of two bromine atoms and a hydroxyl (R is ribose or deoxyribose residue).
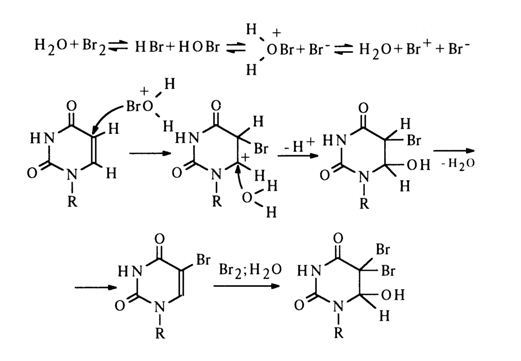
The reaction rates differ from one step to another, and the 5-bromo-6hydroxy-6,6-dihydro derivative can also be obtained with a good yield.
Similarly, a bisulfite ion is added at the C5_C6 double bond. This reaction has been studied especially well on nucleotides.
The addition of osmium tetraoxide at the C5-C6 double bond causes hydroxylation of the pyrimidine ring.
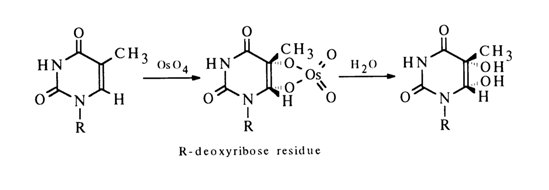
The oxidation of thymidine is much easier than that of uracil and cytosine ribo- and deoxyribonucleosides.
The family of furocumarin derivatives known as psoralens has been actively investigated with regard to the capacity of these compounds to act both as dermal photosensitising agents and as probes
of nucleic acids structure and function. The biological activity of psoralens is primarily the result of covalent binding which involves three discrete steps: (1) nonbonding, intercalating bonding to the DNA; (2) upon irradiation at 365 nm, formation of a monoaddition product between the psoralen and a DNA base, probably a pyrimidine; (3) absorption of a second photon by some monoadducts, which results in interstrand cross-linking. The native DNA were examined. Five nucleoside-HMT monoaddition products were isolated and characterised; three were deoxythymidine adducts and two were deoxyuridine adducts from deoxycytidine-HMT adducts. Two of these adducts are given below:
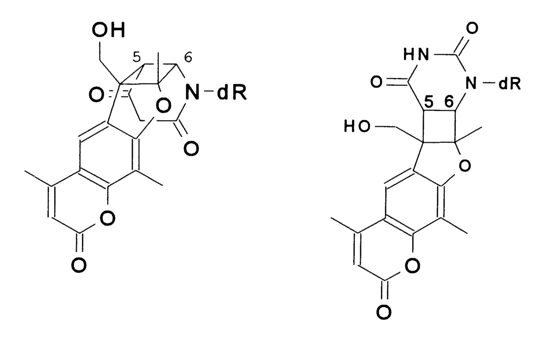
The results of their study indicated that (1) a limited number of nucleosidepsoralen adducts are formed with native dsDNA, and (2) the stereochemistry of the adducts is apparently determined by the geometry of the noncovalent intercalated complex formed by HMT and DNA prior to irradiation.
The double bond in pyrimidine nucleosides lends itself readily to hydrogenation. 5,6-Dihydro derivatives are obtained with a good yield in catalytic hydrogenation in the presence of rhodium on aluminium oxide and also when platinum or palladium catalysts are employed.
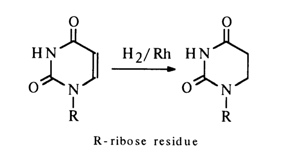
In the case of cytidine, the reaction does not stop at the step of addition of one mole of hydrogen, which is followed by a subsequent reduction step. Therefore, when 5,6-dihydrocytidine is the desired product, the process must be carefully controlled. Reduction of pyrimidine nucleosides is also possible using sodium in liquid ammonia or a sodium amalgam in water, however, complex mixtures of reduction products are formed in such cases.
Purine nucleosides are immune to the action of the above-mentioned reducing agents.
2.1.6 Reactions Involving Exocyclic Amino Groups
From the standpoint of amino group properties, cytosine, adenine and guanine closely resemble aromatic amines containing electron-acceptor substituents (e. g., para-nitroaniline) in their rings. The pKa values of the amino groups of the above heterocycles range from 2.5 to 4. None the less, reactions with electrophilic reagents proceed readily at the amino groups of the bases. They are universally used in the synthesis of various derivatives of nucleosides and for their modification at the monomer and polymer levels. The most typical reaction in this category is amino group acylation (the acylation also involving the hydroxy groups of the carbohydrate moiety). Of the three major nucleosides containing amino groups in the heterocyclic nucleus, cytidine enters into acylation reactions most easily. For selective N-acetylation the reaction is carried out in an alcohol solution with boiling in the presence of an equimolar amount of acetic anhydride.
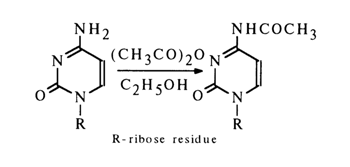
The structure of the acetylation products has been established by UV, IR and NMR spectroscopy. When the reaction is conducted under vigorous conditions (surplus of the acylating agent, high temperature), the acylation involves not only the amino group of the heterocycle but also the heterocyclic nitrogen (to say nothing of the hydroxy groups of the carbohydrate moiety), for example:
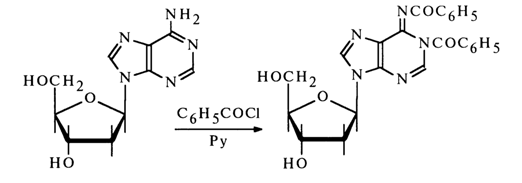
Benzoylation of deoxyguanosine (and guanosine) under the same conditions gives a similar derivative, for example:
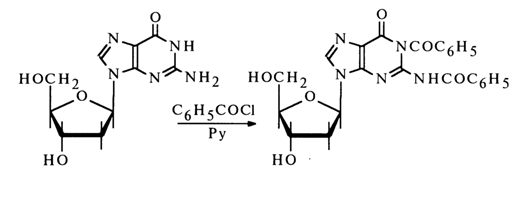
An important reaction also involving the amino group is that between respective nucleosides and aldehydes. A reaction with formaldehyde in aqueous solutions is most likely to yield N-methylol derivatives of RNHCH2OH. Only guanosine and its derivatives can enter into a reaction with glyoxal:
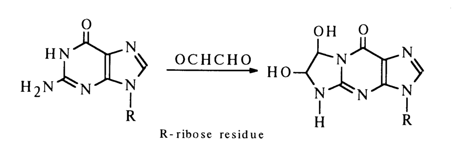
The selectivity of this reaction indicates how important the -NH-C(NH2)=N- group is for its course. 3-Ethoxybutan-2-on-l-al reacts just as guanine nucleosides do:
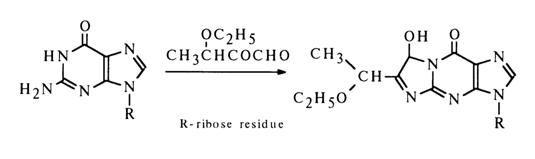
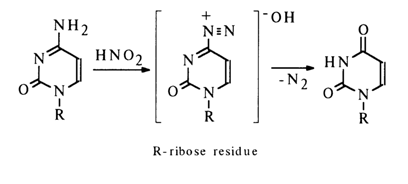
Nucleosides containing amino groups in the heterocycle typically enter into a reaction with nitrous acid. A case in point is adenosine which is converted into inosine in the presence of nitrous acid:
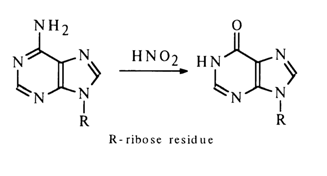
Deamination of guanosine and cytidine yields, respectively, xanthosine and uridine. The reaction seems to result in the corresponding diazonium salt in which no delocalization of the positive charge takes place and which readily dissociates, just as diazoalkyls, in an aqueous medium giving a hydroxy derivative. For instance: