![]() Go to frame view (Recommended only for
screen resolution 1024x768)
Go to frame view (Recommended only for
screen resolution 1024x768)
6.7 Determination of Nucleotide Sequences in RNA
The determination of the primary structure of RNA is the first step in elucidating its functional capabilities. The earliest approach used to sequence rather large polynucleotides was the so-called block method developed in the mid sixties for transfer RNAs. The method is based on a combination of complete and partial hydrolyses of tRNAs with pyrimidyl and guanyl RNases followed by isolation of oligonucleotides and their sequencing. By obtaining overlapping blocks one can reconstruct the initial molecular structure. This approach has been treated at length elsewhere. Since this is all history now, we shall not dwell on this method at all but discuss the fast RNA sequencing techniques extensively used at present. It would be natural to apply the chemical degradation and polymerase copying procedures to RNAs. As a matter of fact, both methods started being developed at the same time (1977-80) and, in spite of the difficulties stemming from the different structures of DNA and RNA, RNA sequencing techniques similar to those for DNA have been elaborated.
6.7.1 Direct Chemical Sequencing of RNAs Labeled at the 3'-End
RNA sequencing based on statistical chemical degradation of the polyribonucleotide chain is identical with the method described above for DNA. True, attempts to create adequate conditions for
b-elimination stumbled against a serious difficulty arising from the presence of a 2'-hydroxyl group in RNA monomers, or ribonucleotides, and the associated high lability of the internucleotide linkages in the presence of bases.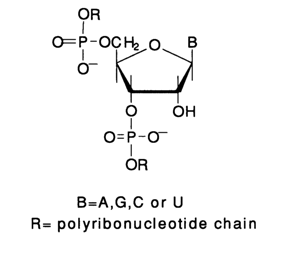
This is why it was impossible to use piperidine (pKa 11.2) which would lead to non-specific cleavage of any internucleotide linkage.

Consequently, different conditions for
b-elimination had to be provided. To this end, piperidine was replaced by a weaker base, namely, aniline (PK 4.5). Besides, the presence of a 2'-hydroxyl affects the b-elimination process itsell The phosphate group is easily eliminated from the 3' position of the sugar moiety, deprived of the heterocyclic base, which is most probably due to the sufficient mobility of the a-proton. As a consequence, the polyribonucleotide chain is cleaved at the site of the removed base. However, the modified sugar moiety in the emerging polyribonucleotide fragment contains a methylene group (as a result of enol-to-ketone conversion) at the former 3' position of ribose. In such a compound (as well as after its conversion into a Schiffs base), the proton can be removed only from the a-position or, in other words, from the metyhlene group, but not from the P-position (former 4' position of ribose). Consequently, the second phosphate group is not removed (from the former 5' position of the sugar), and at their 3' end the 5' fragments of the polyribonucleotide contain, after such transformations, an esterified phosphate rather than a free phosphate group (as in the case of DNA). Therewith, the former ribose, which is essentially an a-diketone derivative, may undergo different transformations. Naturally, this may affect the band pattern after PAGE if 5'-labeled RNA is used for analysis. This is precisely why RNA labeling prior to sequencing at the 3' end (see below) has become extremely popular. In this case, RNA fragments have identical monophosphate groups at the 5' end.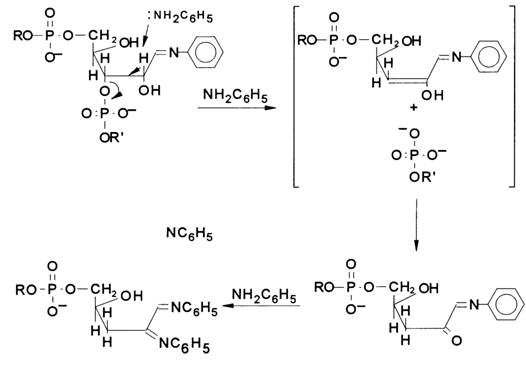
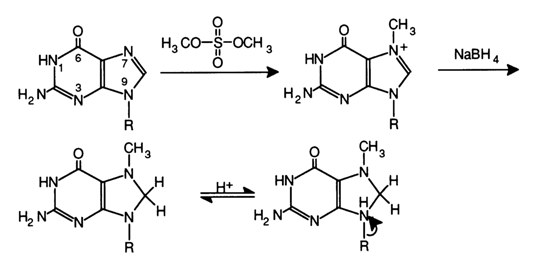
Fig 6-23. Methylation of the guanine at N7 with subsequent reduction with sodium borohydride.
The methods for modifying heterocyclic bases, used in the context of DNA, have undergone some changes, when applied to RNA, due to the pronounced lability of the internucleotide linkages.
Guanine Reaction. The modification of guanines in RNA, as well as in DNA, is performed with the aid of dimethyl sulfate (see Fig. 6-23). In this case, the methylation involves predominantly the nitrogen at position 7. However, such an N7-methylated guanosine link in RNA is stable at neutral pH values. Therefore, for guanine to be removed, the double bond in the imidazole ring is reduced with sodium borohydride (the reduction proceeds easily under rather mild conditions), and the glycosidic bond in the resulting dihydro derivative of 7-methylguanosine becomes highly labile and easily hydrolysed when treated with aniline. The limited methylation of RNA with dimethyl sulfate followed by reduction with NaBH4 and aniline hydrolysis yields polyribonucleotide fragments specifically cleaved at G.
Adenine Reaction (A > G). When diethyl pyrocarbonate reacts with nucleic acids in aqueous solutions, the purine base nitrogens undergo carboethoxylation. It is primarily the nitrogens at position 7 of adenine and guanine that are involved in this case, and the imidazole ring is opened. As a result, the chain can be cleaved with aniline at that position. Since the reaction at adenines proceeds at a rate seven times faster than in the case of guanines, it is used to determine the A positions in polyribonucleotides (see Fig. 6-24).
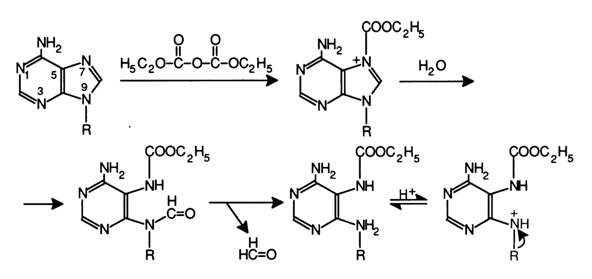
Fig. 6-24. Reaction of diethyl pyrocarbonate with adenine in RNA, which leads to opening of the imidazole ring.

Fig. 6-25. Hydrazinolysis of uracils in RNA, resulting in a labile N-glycosidic bond broken after treatment with aniline.
Uracil Reaction. The mechanism by which unprotonated molecules of hydrazine react with uridine has already been described in the context of cytidine and thymidine. The rate of this reaction, when uracil is involved, is much faster than in the case of C and T (U >> C > T). Once the pyrazolone ring is formed (see Fig. 6-25), it is readily detached from the glycosidic carbon, and an attack with aniline yields a Schiff's base, which leads to
b-elimination of the 3'-phosphate group and, consequently, cleavage of the RNA chain at that position.Cytosine Reaction (C > U). Anhydrous hydrazine reacts primarily at cytosines in the presence of 3 M NACI Oust as in the case of DNA, the reaction is predominantly at C in the presence of 2 M NACI). The mechanism of the reaction at cytosines has already been described.
Figure 6-26 shows schematically the chemical degradation of a polyribonucleotide at the position where the above-described modification of a particular base has taken place in the presence of aniline. The glycosidic hydroxyl resulting from hydrolysis of the N-glycosidic bond (at the modified base site) gives rise to a tautomeric aldehyde form of the sugar moiety. This form reacts with aniline to yield an aldimine (Schiff's base), and, as has already been mentioned,
b-elimination and, consequently, RNA cleavage takes place. Aniline titrated with acetic acid to pH 4.6 ensures an optimal pH value for b-elimination and sufficiently mild conditions to avoid nonspecific cleavage of internucleotide linkages in RNA.As has already been pointed out, for the sequencing results to be unambiguous, the label is usually incorporated into the 3' end of RNA. This is achieved through a reaction between RNA and [5'-32P]pCp in the presence of RNA ligase:
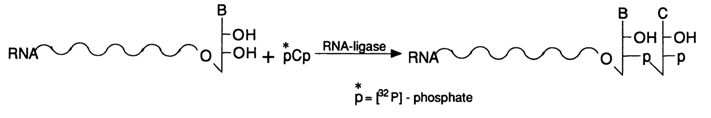
Such an RNA labeled at the 3' end is separated by PAGE then sequenced.
However, the 3' end may also be labeled after the chemical modifications. This saves a lot of time when several RNAs have to be sequenced in the same experiment. For example, as can be seen from Figure 6-27, four chemical reactions may be conducted with a mixture of several labeled RNAs (RNAs may also be labeled after the chemical modification), followed by fractionation in polyacrylamide gel, elution, and pelleting of an individual RNA.
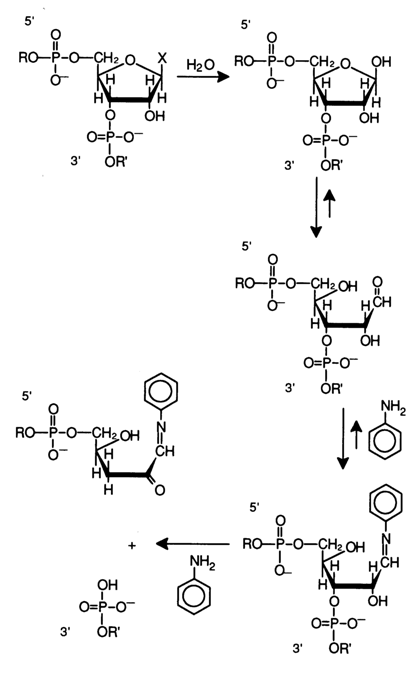
Fig. 6-26. Degradation of RNA at modified bases, yielding a set of 3'-terminal fragments with 5'-phosphate.
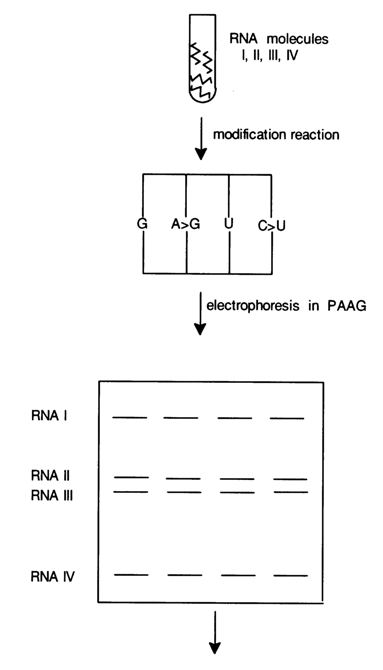
Fig. 6-27. Simultaneous sequencing of several RNAs.
Each modified RNA separated in this fashion is treated with aniline (with cleavage of the polynucleotide chain), then subjected to PAGE. Figure 6-27 shows that RNA molecules I, II, III and IV are separated by PAGE after restricted chemical modification (at G, A > G, C > U and U). As a result, only four chemical reactions are conducted instead of sixteen. The aniline treatment is carried out immediately after the elution from polyacrylamide gel and pelleting. The pellets are then applied onto a standard gel to be subjected to electrophoresis and autoradiographed.
Figure 6-28 is an autoradiogram of the sequencing gel illustrating the primary structure of 5S RNA (the structure is read from the 121st nucleotide; i.e., from the 3' end). The gel electrophoresis was performed under standard conditions in the presence of 7 M urea.

Fig. 6-28. Autoradiogram of the sequencing gel of 5S RNA labeled at the 3'end with the aid of [5'-32P]pCp and RNA ligase.
6.7.2 Direct Enzymatic Sequencing of RNA
A modification of direct RNA sequencing is the enzymatic method of statistical cleavage of internucleotide linkages with the aid of RNases selectively breaking the phosphodiester bonds formed by pyrimidine and purine nucleoside 3'-phosphates.
As was pointed out in the introduction, the presence of RNases A (pyrimidyl RNase) and T1 (guanyl RNase) whose substrate specificity has been carefully elucidated has made it possible to develop the block method of RNA sequencing, used already in the mid sixties for determining the sequence of rather large molecules of individual RNAs. These RNases, plus some new ones whose substrate specificity will be described in the following, were used for statistical cleavage of polyribonucleotides instead of their chemical degradation. Four enzymes are usually employed for the purpose. The following schemes illustrate the cleavage of internucleotide linkages by each of them.
RNase T, cleaves internucleotide linkages formed by guanosine 3'-phosphates in the polyribonucleotide:
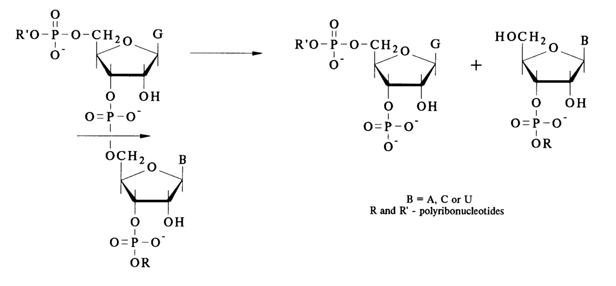
Hence, RNase T, serves as a means for determining the position of G during RNA sequencing.
RNase U2: The specificity of this enzyme is similar to that of the
previous one with the difference that this RNase cleaves internucleotide linkages formed
by adenosine 3'-phosphate and is used to determine the position of A in the RNA: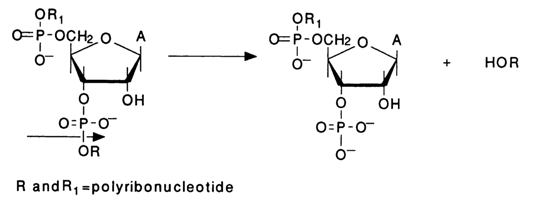
RNase Phv M (from Phyzarium polycephalum) cleaves
internucleotide linkages by the same mechanisms ![]() but involves bonds of two types: thoses formed by adenosine 3'-
and uridine 3'-phosphates:
but involves bonds of two types: thoses formed by adenosine 3'-
and uridine 3'-phosphates:
Ap Æ N UpÆN
RNase BC (from Bacillus cereus) hydrolyses internucleotide linkages formed by pyrimidine nucleoside 3'-phosphates similarly to RNase A (pyrimidyl RNase):
Up Æ N Cp Æ N
Like in the previous cases, the emerging 3'-terminal RNA fragments lack phosphate groups at the 5' end (in contrast to the 3'-terminal fragments resulting from chemical degradation of RNA). In addition to the main ribonucleases use is also made of RNase CL3 isolated from chicken liver. This enzyme is highly specific, digesting primarily internucleotide linkages formed by cytidine 3' -phosphates: CpÆ N. There is yet another, less specific ribonuclease used for RNA sequencing, namely, nuclease M, hydrolysing linkages NÆ pA, NÆ pU and NÆ pG. In this case, the 3'-terminal RNA fragments already have phosphate groups at the 5' end.
All reactions in which RNAs are digested by these enzymes usually take 12 minutes at 550C with subsequent application onto polyacrylamide gel and electrophoresis. Here, just as in direct chemical degradation, 3'-labeled RNA is used. However, during enzymatic digestion the 5' end of RNA may also be labeled with the aid of polynucleotide kinase and [
g-32P] ATP, because all 5' terminal fragments resulting from statistical enzymatic hydrolysis with RNases Tl, U2, Phv M, BC and CL3 have a 3'-terminal 2',3-cyclic phosphate group. Statistical hydrolysis of 3 mg RNA under standard conditions requires a single activity unit from each enzyme. Along with enzymatic digestion, which makes it possible to identify the positions of the four nucleotides along the RNA chain from the 5' or 3' end, it has also become common practice to resort to incomplete alkaline hydrolysis of the RNA under analysis. Application of alkaline hydrolysis (the autoradiogram usually shows all RNA fragments differing by a single nucleotide) simplifies the reading of the gel. Figures 6-29 and 6-30 are autoradiograms of sequencing gels after sequencing of E. coli 5S RNA with the aid of enzymes produced by "Pharmacia". The only difference in the sequencing procedures was use of 5'-labeled RNA (Fig. 6-29) and 3' labeled RNA (Fig. 6-30).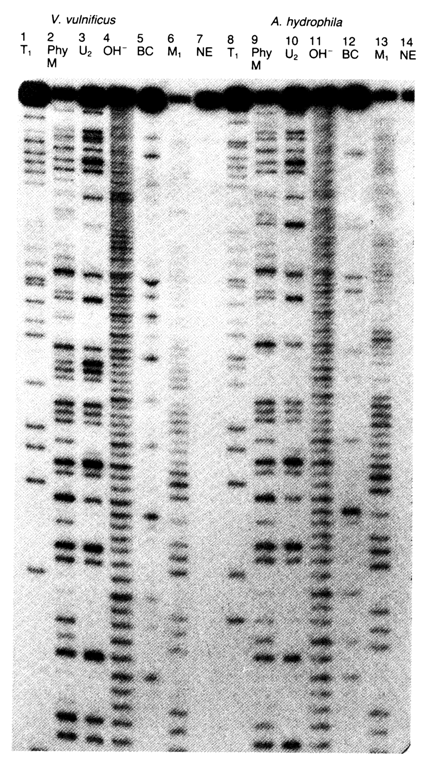
Fig. 6-29. Autoradiogram of the sequencing gel of E. coli 5S RNA labeled at the 5' end. 20 % polyacrylamide gel containing 7M urea is used. Channel 1 NE is control (without enzyme). Channel OH corresponds to alkaline hydrolysate of E. coli 5S RNA. The other channels correspond to hydrolysates involving the above-described enzymes. Channels 10-12 correspond to hydrolysates involving RNase V1 isolated from cobra venom and cleaving RNA only over double-stranded portions.
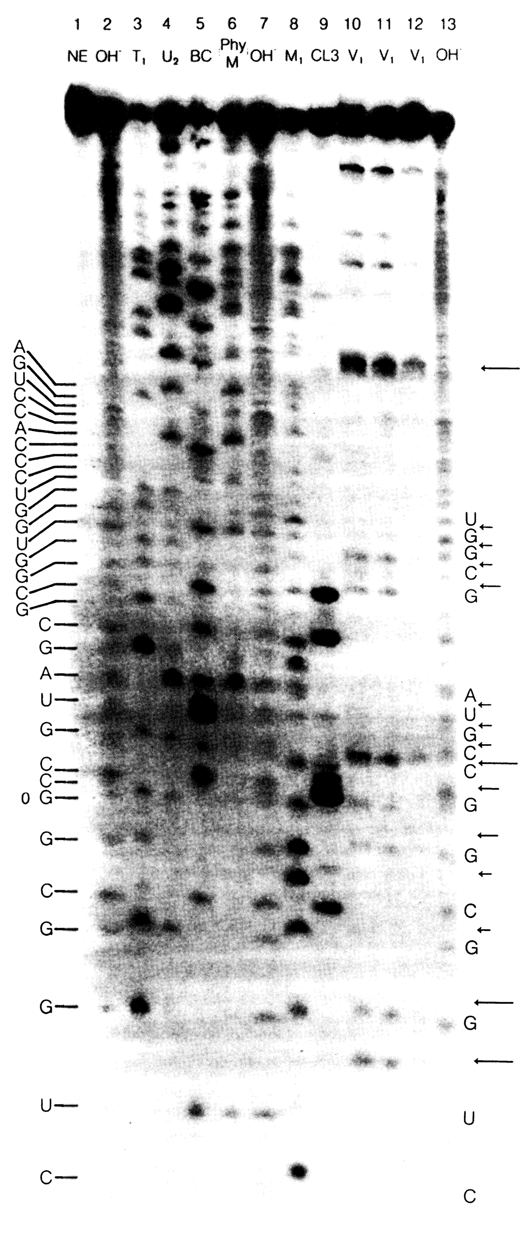
Fig. 6-30. Autoradiogram of the sequencing gel of E. coli 5S RNA labeled at the 3' end with the aid of [5'-32P]pCp and RNA ligase. The sequencing conditions and channels are the same as in Fig. 6-29.
6.7.3 Sequencing of High-Molecular Weight RNAs by Polymerase Copying with the Aid of Reverse Transcriptase
The possibility of DNA copying by way of reverse transcription is widely used for sequencing high-molecular weight RNAs. Two main procedures of RNA sequencing in this manner exist. The first procedure is based on obtaining a single-stranded cDNA with separation in polyacrylamide gel before its direct sequencing. The other calls for obtaining a double-stranded cDNA through reverse transcription and cloning in a suitable vector with subsequent sequencing by any method.
Since the RNA sequencing method described here ultimately boils down to DNA sequencing, we will consider a few examples illustrating only procedures for obtaining individual cDNAs.
The first procedure was used to sequence interferon mRNA. In order to obtain a transcript of interferon mRNA, the latter was not separated from a mixture of polyadenylated mRNAs isolated from human fibroblasts. Instead a primer was added to the complex mixture of mRNA, namely synthetic 15membered oligodeoxyribonucleotide complementary with respect to the 3' terminal portion of the interferon mRNA. The nucleotide sequence of the primer, which was expected to produce a complementary complex only with the interferon mRNA, was determined from the known initial structure of the C-terminal region of the protein - fibroblast interferon to be precise. In this case, account is taken of the codons from human genes used predominantly for polycodon amino acids. The primer engineering procedure is shown in below.
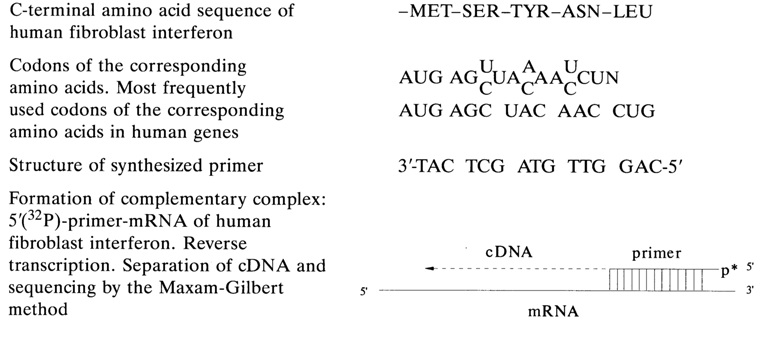
After synthesis, 5'-terminal phosphate is inserted into the 15-linked oligonucleotide with the aid of [
g-32P] ATP and polynucleotide kinase. The labeled primer forms a complementary complex only with interferon MRNA. Following incubation with reverse transcriptase, cDNA is separated by PAGE and sequenced by the Maxam-Gilbert method.The use of synthetic primers opens up wide possibilities for direct sequencing of rare mRNAs without their separation from complex mRNA mixtures. It also helps to eliminate the highly laborious procedure of mRNA isolation and purification as well as cloning. Among the disadvantages of the method is prefact that only a sequence of 250 to 300 nucleotides can be determined and the fact that structure can be determined only from a single DNA strand. As a matter of fact, the first difficulty is easily circumvented at present by creating new primers in the course of sequencing (it takes a maximum of one day to synthesize and purify a 15-unit oligonucleotide), which are complementary to the RNA portions whose primary structure is already established.
The other approach to RNA sequencing is based on the synthesis of a double-stranded DNA copy. This is how the primary structure of the influenza virus RNA was established. Figure 6-31 shows schematically the transformation of the influenza virus mRNA to a double-stranded transcript, aided by a synthetic primer. After the double-stranded DNA copy had been treated with nuclease S,, which digests single-stranded portions of DNA, and also with restriction endonucleases, fragments of the RNA transcript were cloned and sequenced by the Sanger method. The successful implementation of this RNA sequencing approach is associated primarily with automation of oligodeoxynucleotide synthesis. Synthetic oligonucleotides are employed not only as primers during reverse transcription but also as probes in identification of RNA and DNA.
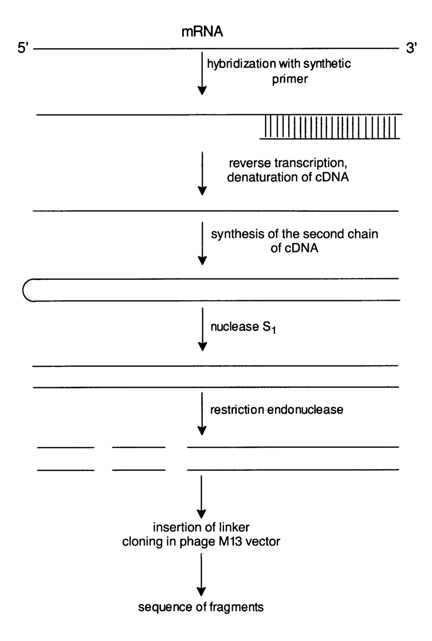
Fig. 6-31. Cloning of fragments of a double-stranded DNA produced by reverse transcription of mRNA and synthetic priming, suitable for sequencing.